
MRM Insights: Pathomechanisms in corner fracture type spondylometaphyseal dysplasia

Philippe Campeau

Neha Dinesh

Dieter Reinhardt
Every month, in MRM Insights, a member of the MRM Network is writing about stem cells and regenerative medicine from a different perspective. This month, Dieter Reinhardt, Professor in the Department of Anatomy and Cell Biology at McGill University, Dr. Philippe Campeau, Assistant Clinical Professor in the Department of Pediatrics at Université de Montréal and Adjunct Professor at McGill, and Neha Dinesh, Ph.D. Candidate in their Lab, are discussing pathomechanisms in corner fracture type spondylometaphyseal dysplasia.
Investigating the pathomechanisms in rare corner fracture type spondylometaphyseal dysplasia caused by fibronectin mutations – an iPSC-based approach
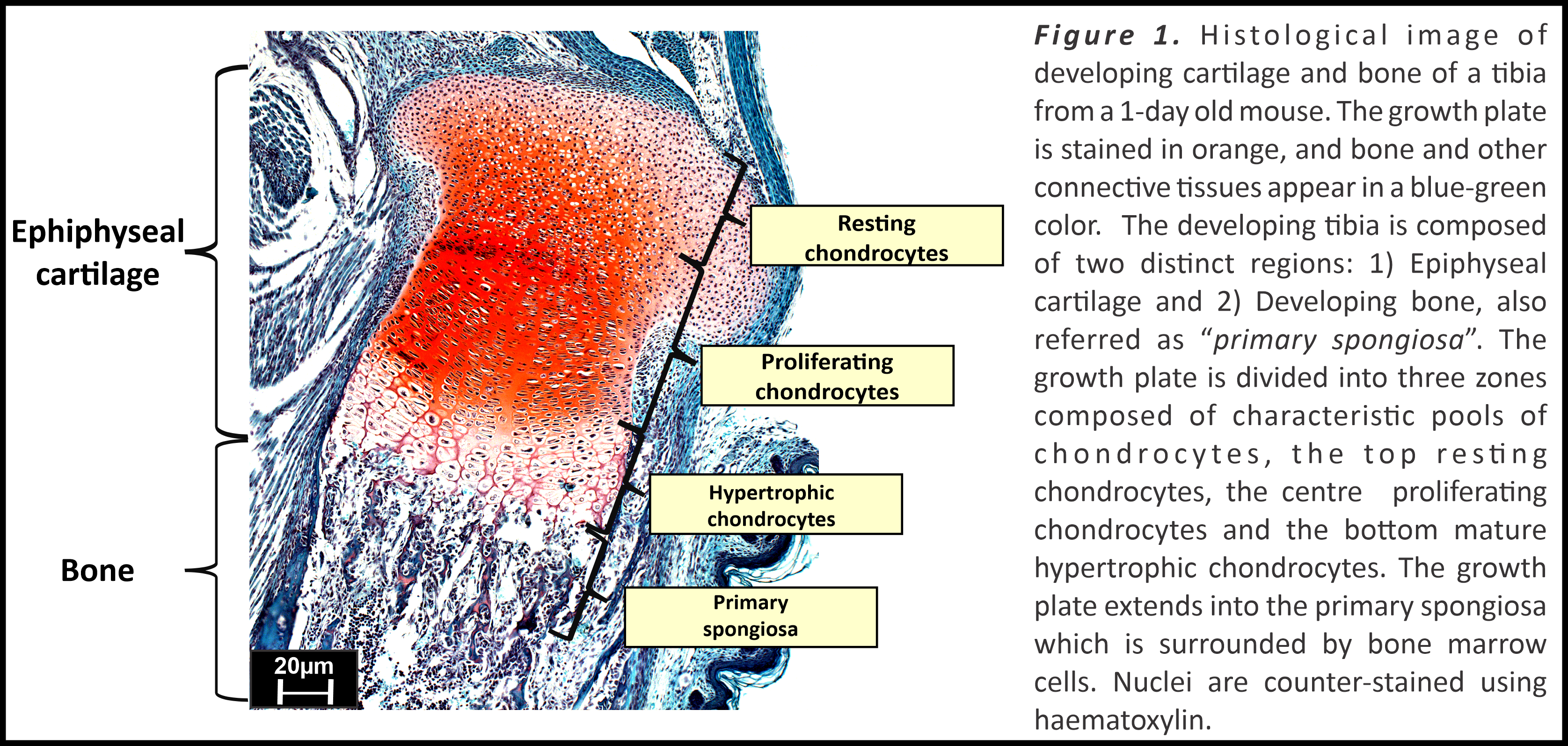
Somatic stem cells are the building blocks of tissue development and homeostasis based on their hallmark properties of multipotency and the ability to undergo self-renewal [1]. Differentiation of stem cells into specific cell types in various tissues are regulated by several intrinsic and extrinsic factors, such as transcription factors, growth factors and interaction with extracellular matrix (ECM) proteins. The skeletal stem cells possess trilineage potential to differentiate into chondrocytes, osteoblasts or marrow stromal cells based on the tissue microenvironment and developmental stage [2,3]. Skeletal development originates during embryogenesis with condensation of mesenchymal stem cells (MSCs) in developing limb buds and differentiation into chondrocytes, leading to the formation of the growth plate (Figure 1). The chondrocytes in the embryonic cartilage template are first resting chondrocytes which then differentiate into collagen type II expressing proliferating chondrocytes. These cells further differentiate to express collagen type X and matrix metalloprotease 13 and then become mature hypertrophic chondrocytes that lay down the initial mineralized bone matrix [4]. A fraction of these chondrocytes continues to differentiate into osteoblasts and bone marrow stromal cells [5], facilitating regulated skeletal development. Growth plate stem cells hence are responsible for development and maintenance of the structural integrity of the growth plate.
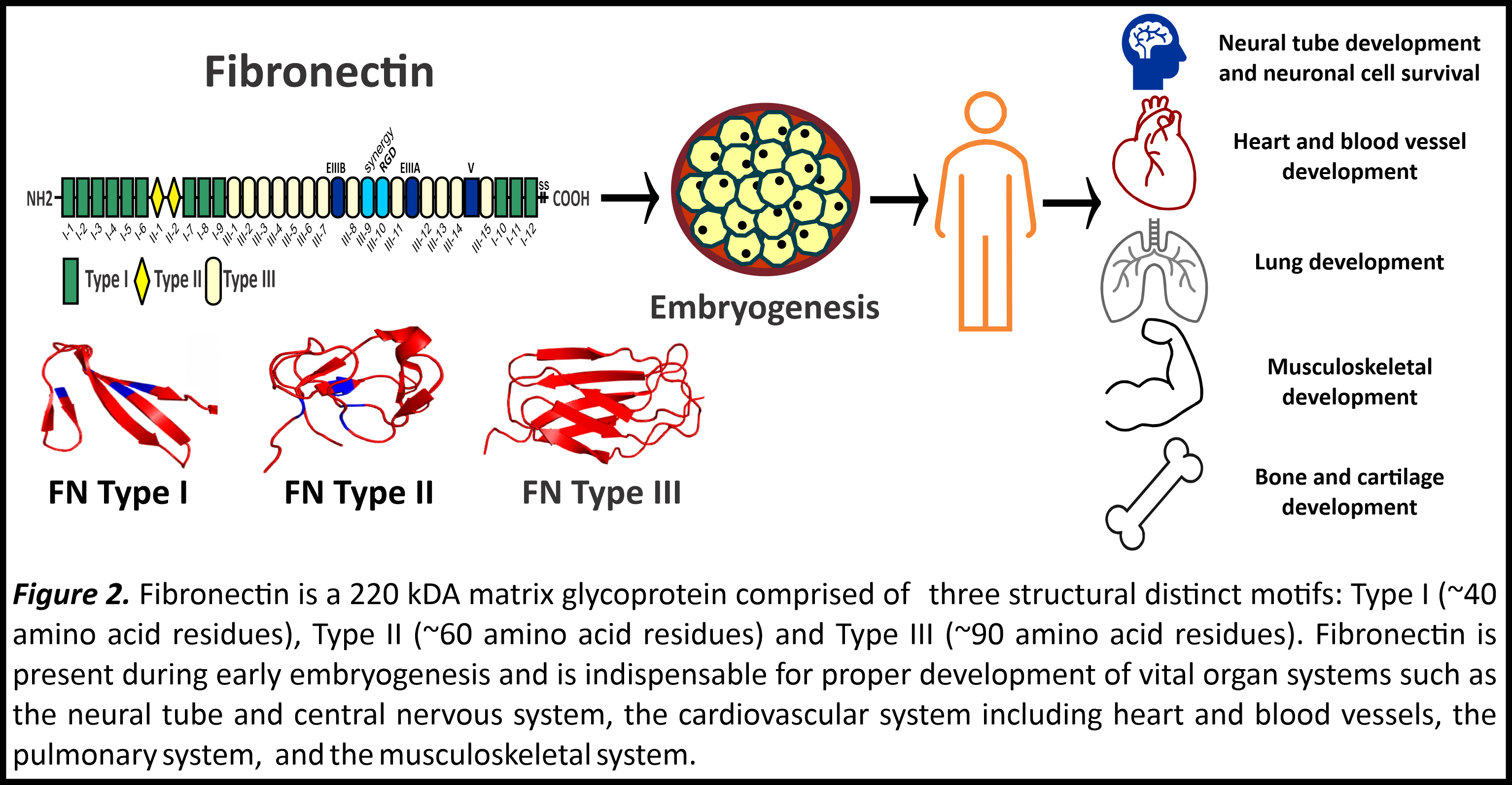
Fibronectin is a master organizing glycoprotein in the ECM that not only provides a framework for assembly of other matrix proteins, but also facilitates stem cell function, proliferation, and differentiation [6]. Fibronectin is secreted from cells as a 440 kDa compact dimeric protein. Its interaction with cells via integrin cell receptors (e.g. α5β1 integrin) initiates changes in the actin cytoskeleton facilitating extension of the compact dimer followed by fibronectin fiber assembly. Research throughout decades has established the physiological importance of fibronectin in the development of several vertebrate organ systems (Figure 2) [7]. Absence of fibronectin during embryogenesis leads to lethality caused by failure of the neural tube and cardiovascular development and absence of limb bud formation [7]. Fibronectin is vital for mesenchymal condensation [6], and knockdown of fibronectin impairs the differentiation of MSCs in cell culture [8]. It is well demonstrated by several studies that fibronectin promotes stem cell differentiation into chondrocytes and osteoblasts [9-11]. However, evidence of skeletal pathologies linked to mutations in fibronectin were not identified until 2017, when the Campeau and Reinhardt laboratories demonstrated for the first time that heterozygous mutations in the fibronectin gene (FN1) causes “corner fracture” type spondylometaphyseal dysplasia (CF-SMD) [12]. Our research groups identified initially six fibronectin mutations in CF-SMD patients from seven different families. These fibronectin mutations, when recombinantly expressed in human embryonic kidney cells (HEK293), displayed impaired protein secretion, meaning that the mutations prevented secretion of fibronectin from cells. The number of FN1 mutations leading to CF-SMD were further expanded by two additional follow-up studies [13,14].
CF-SMD represents a subgroup of skeletal dysplasias, which are rare heterogenous disorders, affecting primarily the spine and growth plate with a combined prevalence of 1 in 5000 births [15]. The identified patients with CF-SMD displayed significant skeletal abnormalities including dwarfism, scoliosis, coxa vara, presence of ovoid vertebral bodies and irregular ossification in the metaphyses that appears as corner fractures in radiographs. Most of these patients underwent surgery for scoliosis repair. It is interesting to note that mutations in several other matrix proteins such as collagen type I, II, X, IX and XI [16-18], and cartilage oligomeric matrix protein (COMP) [19] also cause other forms of SMD. In addition, the molecular phenotype observed for collagen type X or COMP mutations overlap with fibronectin mutations, as they also cause intracellular retention of the mutant proteins [20,21].
One of the primary goals of our research groups is to obtain pathomechanistic insights into how fibronectin mutations lead to CF-SMD. We have established induced pluripotent stem cells (iPSC) with skin fibroblasts donated by CF-SMD patients. iPSCs are an excellent tool for modelling and studying both physiological as well as pathological mechanisms. Our working model involves differentiation of iPSCs into MSCs and then into chondrocytes, which enables us to define alterations in chondrogenic pathways imposed by mutant fibronectin. From our previously published work [13,14], it is evident that these CF-SMD causing fibronectin mutations may affect stem cell functions and their differentiation via two possible pathways. One possibility is by intracellular accumulation of this matrix protein which may lead to cellular stress, another possibility is through deficiency of fibronectin in the microenvironment of cells. It was previously demonstrated that a fibronectin deficient matrix can impair MSC differentiation [8]. In addition, intracellular protein accumulation is also known to cause skeletal dysplasias as observed for mutations in SLC26A2 [22,23], collagen type X and COMP [19]. In our work with patient iPSCs we analyze and evaluate how the consequences of fibronectin mutations could be translating into CF-SMD in the affected individuals.
Presently, only very limited therapeutic treatments are available for skeletal dysplasia patients which are clinically tested and approved. Stem cell transplantation, growth hormone treatment and use of synthetic peptides such as C-type natriuretic peptides (achondroplasia) are few of the clinical treatments that are available today [24]. With our ongoing research work to understand the pathomechanisms of fibronectin mutations in CF-SMD, we hope to get a better insight of how fibronectin directs stem cell differentiation into chondrocytes at the molecular level and how mutations in fibronectin alter cell function and differentiation. We hope that our findings will contribute in the future towards identifying pharmacologically targetable pathways in treating CF-SMD and related genetic disorders.
About the authors
Dieter P. Reinhardt, Ph.D. is a Principal Investigator and Distinguished James McGill Professor in the Faculty of Medicine and Health Sciences and the Faculty of Dental Medicine and Oral Health Sciences at McGill University. His research focusses on determining the mechanisms of biogenesis and function of extracellular fiber systems under normal and pathological conditions.
Dr. Philippe Campeau, M.D. is a Clinical Associate Professor at the University of Montreal and a Principal Investigator at the CHU Sainte-Justine. His research work primarily involves the study of epigenetic diseases, skeletal dysplasias, identifying disease-causing genes, and improving the management of children affected by these conditions.
Neha Dinesh, M.Sc. is a Ph.D. student in the Department of Anatomy and Cell Biology at McGill University and is co-supervised by Profs. Reinhardt and Campeau. Her research project involves investigating the consequence of fibronectin mutations in skeletal development and in spondylometaphyseal dysplasia.
References
1. Tweedell KS. (2017) The adaptability of somatic stem cells: A review. J Stem Cells Regen Med 13, 3-13
2. Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, Lu WJ, Senarath-Yapa K, Chung MT, Marecic O, Tran M, Yan KS, Upton R, Walmsley GG, Lee AS, Sahoo D, Kuo CJ, Weissman IL, Longaker MT. (2015) Identification and specification of the mouse skeletal stem cell. Cell 160, 285-298
3. Matsushita Y, Ono W, Ono N. (2020) Skeletal stem cells for bone development and repair: Diversity matters. Curr Osteoporos Rep 18, 189-198
4. Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. (2008) Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 40, 46-62
5. Ono N, Ono W, Nagasawa T, Kronenberg HM. (2014) A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol 16, 1157-1167
6. Burton-Wurster N, Lust G, MacLeod JN. (1997) Cartilage fibronectin isoforms: In search of functions for a special population of matrix glycoproteins. Matrix Biol 15, 441-454
7. de Almeida PG, Pinheiro GG, Nunes AM, Goncalves AB, Thorsteinsdottir S. (2016) Fibronectin assembly during early embryo development: A versatile communication system between cells and tissues. Dev Dyn 245, 520-535
8. White ES, Muro AF. (2011) Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 63, 538-546
9. Zhang H, Chen X, Xue P, Ma X, Li J, Zhang J. (2021) FN1 promotes chondrocyte differentiation and collagen production via TGF-beta/PI3K/Akt pathway in mice with femoral fracture. Gene 769, 145-253
10. Casanova MR, Reis RL, Martins A, Neves NM. (2020) Fibronectin bound to a fibrous substrate has chondrogenic induction properties. Biomacromolecules 21, 1368-1378
11. Moursi AM, Damsky CH, Lull J, Zimmerman D, Doty SB, Aota S, Globus RK. (1996) Fibronectin regulates calvarial osteoblast differentiation. J Cell Sci 109, 1369-1380
12. Lee CS, Fu H, Baratang N, Rousseau J, Kumra H, Sutton VR, Niceta M, Ciolfi A, Yamamoto G, Bertola D, Marcelis CL, Lugtenberg D, Bartuli A, Kim C, Hoover-Fong J, Sobreira N, Pauli R, Bacino C, Krakow D, Parboosingh J, Yap P, Kariminejad A, McDonald MT, Aracena MI, Lausch E, Unger S, Superti-Furga A, Lu JT, Baylor-Hopkins Center for Mendelian Genomics, Cohn DH, Tartaglia M, Lee BH, *Reinhardt DP, *Campeau PM. *Co-senior authors (2017) Mutations in fibronectin cause a subtype of spondylometaphyseal dysplasia with “corner fractures”. Am J Hum Genet 101, 815-823
13. Cadoff EB, Sheffer R, Wientroub S, Ovadia D, Meiner V, Schwarzbauer JE. (2018) Mechanistic insights into the cellular effects of a novel FN1 variant associated with a spondylometaphyseal dysplasia. Clin Genet 94, 429-437
14. Costantini A, Valta H, Baratang NV, Yap P, Bertola DR, Yamamoto GL, Kim CA, Chen J, Wierenga KJ, Fanning EA, Escobar L, McWalter K, McLaughlin H, Willaert R, Begtrup A, Alm JJ, Reinhardt DP, Makitie O, Campeau PM. (2019) Novel fibronectin mutations and expansion of the phenotype in spondylometaphyseal dysplasia with “corner fractures”. Bone 121, 163-171
15. Marzin P, Cormier-Daire V. (2020) New perspectives on the treatment of skeletal dysplasia. Ther Adv Endocrinol Metab 11, 2042018820904016
16. Walter K, Tansek M, Tobias ES, Ikegawa S, Coucke P, Hyland J, Mortier G, Iwaya T, Nishimura G, Superti-Furga A, Unger S. (2007) COL2A1-related skeletal dysplasias with predominant metaphyseal involvement. Am J Med Genet 143A, 161-167
17. Krakow D. (2015) Skeletal dysplasias. Clin Perinatol 42, 301-319i
18. Gibson BG, Briggs MD. (2016) The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis 11, 86
19. Maddox BK, Keene DR, Sakai LY, Charbonneau NL, Morris NP, Ridgway CC, Boswell BA, Sussman MD, Horton WA, Bachinger HP, Hecht JT. (1997) The fate of cartilage oligomeric matrix protein is determined by the cell type in the case of a novel mutation in pseudoachondroplasia. J Biol Chem 272, 30993-30997
20. Hecht JT, Hayes E, Haynes R, Cole WG. (2005) COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol 23, 525-533
21. Wilson R, Freddi S, Chan D, Cheah KS, Bateman JF. (2005) Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J Biol Chem 280, 15544-15552
22. Galante LL, Schwarzbauer JE. (2007) Requirements for sulfate transport and the diastrophic dysplasia sulfate transporter in fibronectin matrix assembly. J Cell Biol 179, 999-1009
23. Zheng C, Lin X, Xu X, Wang C, Zhou J, Gao B, Fan J, Lu W, Hu Y, Jie Q, Luo Z, Yang L. (2019) Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias. EBioMedicine 40, 695-709
24. Jelin AC, O’Hare E, Blakemore K, Jelin EB, Valle D, Hoover-Fong J. (2017) Skeletal dysplasias: Growing therapy for growing bones. Front Pharmacol 8, 79
Photo Credit: Bright field image of a whole mount skeletal staining of a 1-day old mouse pup, courtesy of Neha Dinesh. Mineralized bone (purple) is stained by alizarin red and cartilaginous skeleton (blue) is stained by alcian blue.