
MRM Insights : Mécanismes pathologiques dans la dysplasie spondylo-métaphysaire type « corner fracture »

Philippe Campeau

Neha Dinesh

Dieter Reinhardt
Chaque mois, dans MRM Insights, un membre du Réseau MRM écrit sur les cellules souches et la médecine régénérative d’un point de vue différent. Ce mois-ci, Dieter Reinhardt, Professeur au Département d’anatomie et biologie cellulaire à l’Université McGill, le Dr Philippe Campeau, Professeur adjoint de clinique au Département de pédiatrie à l’Université de Montréal et Professeur associé à McGill, et Neha Dinesh, Candidate au doctorat dans leur laboratoire, nous parlent des mécanismes pathologiques dans la dysplasie spondylo-métaphysaire type « corner fracture ».
Étude des mécanismes pathologiques dans la dysplasie spondylo-métaphysaire rare de type « corner fracture » causée par des mutations de la fibronectine – une approche basée sur les iPSCs
Les cellules souches somatiques sont les éléments constitutifs du développement tissulaire et de l’homéostasie en fonction de leurs propriétés caractéristiques de multipotence et de leur capacité à se renouveler [1]. La différenciation des cellules souches en types cellulaires spécifiques dans divers tissus est régulée par plusieurs facteurs intrinsèques et extrinsèques, tels que les facteurs de transcription, les facteurs de croissance et l’interaction avec les protéines de la matrice extracellulaire (MEC). Les cellules souches squelettiques possèdent un potentiel de trilignage pour se différencier en chondrocytes, ostéoblastes ou cellules stromales de la moelle en fonction du microenvironnement tissulaire et du stade de développement [2,3]. Le développement du squelette prend naissance au cours de l’embryogenèse avec la condensation des cellules souches mésenchymateuses (CSM) dans le développement des bourgeons des membres et différenciation en chondrocytes, conduisant à la formation de la plaque de croissance (Figure 1). Les chondrocytes dans le gabarit cartilagineux embryonnaire sont d’abord des chondrocytes au repos qui se différencient ensuite en collagène de type II exprimant des chondrocytes proliférants. Ces cellules se différencient davantage pour exprimer le collagène de type X et la métalloprotéase matricielle 13, puis deviennent des chondrocytes hypertrophiques matures qui créent la matrice osseuse minéralisée initiale [4]. Une fraction de ces chondrocytes continue de se différencier en ostéoblastes et en cellules stromales de la moelle osseuse [5], facilitant ainsi le développement régulé du squelette. Les cellules souches de la plaque de croissance sont donc responsables du développement et du maintien de l’intégrité structurelle de la plaque de croissance.
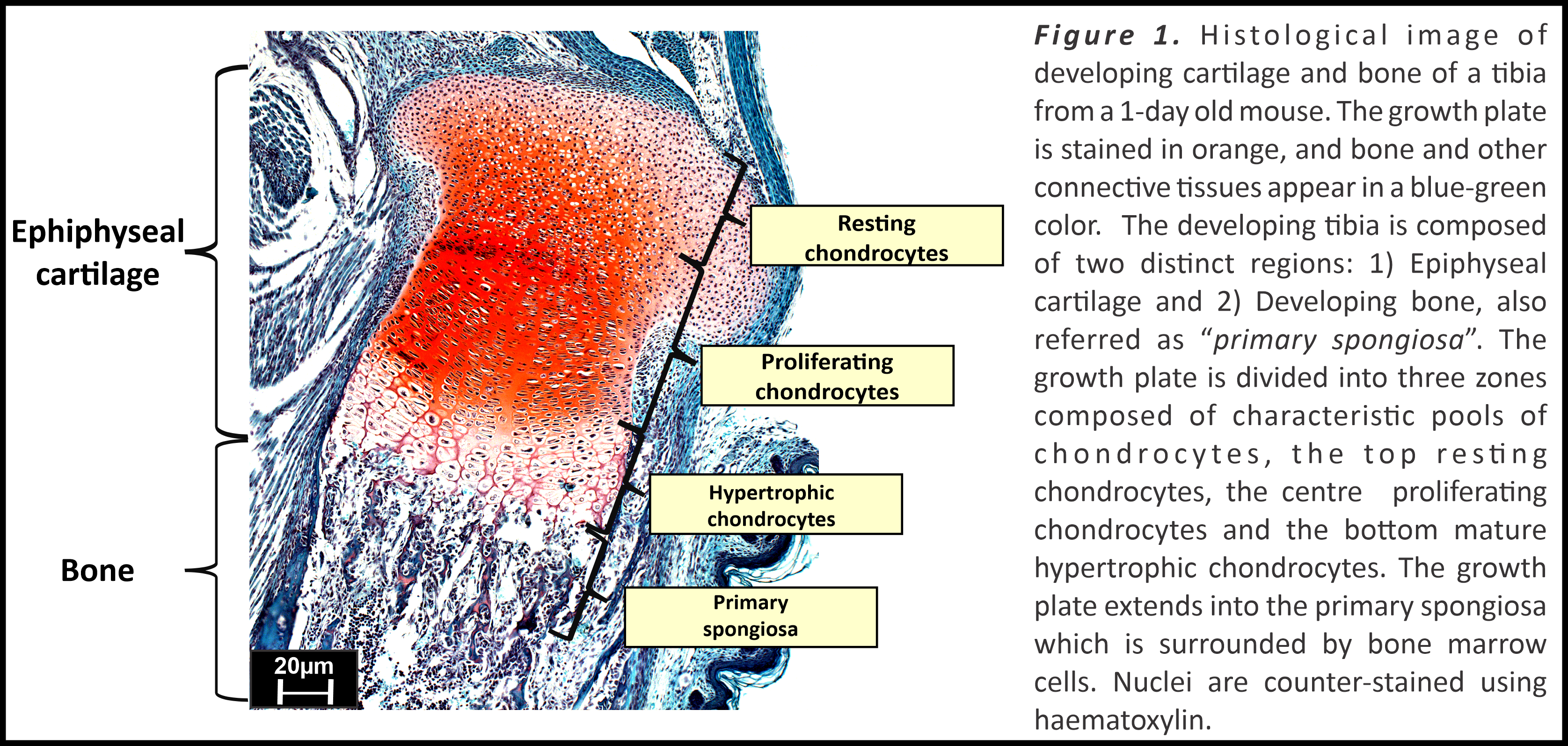
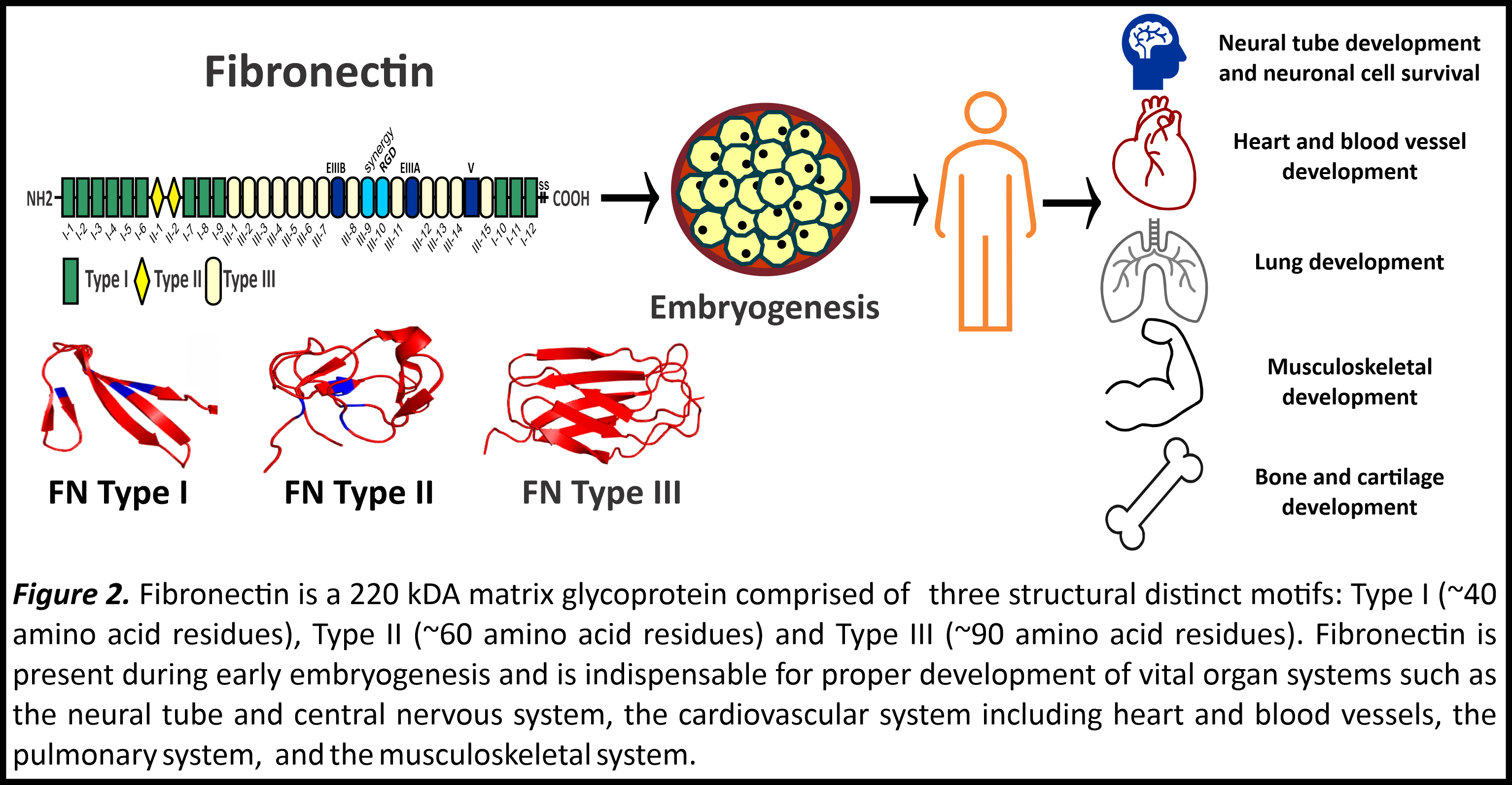
La fibronectine est une glycoprotéine organisatrice maîtresse dans la MEC qui fournit non seulement un cadre pour l’assemblage d’autres protéines matricielles, mais facilite également la fonction, la prolifération et la différenciation des cellules souches [6]. La fibronectine est sécrétée par les cellules sous la forme d’une protéine dimérique compacte de 440 kDa. Son interaction avec les cellules via les récepteurs des cellules d’intégrine (par exemple l’intégrine α5β1) initie des changements dans le cytosquelette d’actine facilitant l’extension du dimère compact suivi d’un assemblage de fibres de fibronectine. Au fil des décennies, la recherche a établi l’importance physiologique de la fibronectine dans le développement de plusieurs systèmes d’organes vertébrés (Figure 2) [7]. L’absence de fibronectine au cours de l’embryogenèse entraîne une létalité causée par une défaillance du tube neural et du développement cardiovasculaire et l’absence de formation de bourgeons des membres [7]. La fibronectine est vitale pour la condensation mésenchymateuse [6], et l’élimination de la fibronectine nuit à la différenciation des CSM en culture cellulaire [8]. Il a été démontré par plusieurs études que la fibronectine favorise la différenciation des cellules souches en chondrocytes et ostéoblastes [9-11]. Cependant, les preuves de pathologies squelettiques liées à des mutations de la fibronectine n’ont été identifiées qu’en 2017, lorsque les laboratoires Campeau et Reinhardt ont démontré pour la première fois que les mutations hétérozygotes du gène de la fibronectine (FN1) provoquent une Dysplasie spondylo-métaphysaire type fracture en coins ou « corner fracture » (CF-SMD) [12]. Nos groupes de recherche ont identifié initialement six mutations de la fibronectine chez des patients atteints de MSM-FK de sept familles différentes. Ces mutations de la fibronectine, lorsqu’elles sont exprimées de manière recombinante dans des cellules rénales embryonnaires humaines (HEK293), ont montré une altération de la sécrétion de protéines, ce qui signifie que les mutations empêchaient la sécrétion de fibronectine des cellules. Le nombre de mutations FN1 menant à la CF-SMD a été encore augmenté par deux autres études de suivi [13,14].
La CF-SMD représente un sous-groupe de dysplasies squelettiques, qui sont des troubles hétérogènes rares, affectant principalement la colonne vertébrale et la plaque de croissance avec une prévalence combinée de 1 naissance sur 5000 [15]. Les patients identifiés atteints de CF-SMD présentaient des anomalies squelettiques significatives, notamment un nanisme, une scoliose, une coxa vara, la présence de corps vertébraux ovoïdes et une ossification irrégulière dans les métaphyses qui apparaît comme des fractures de coin dans les radiographies. La plupart de ces patients ont subi une intervention chirurgicale pour la réparation de la scoliose. Il est intéressant de noter que les mutations dans plusieurs autres protéines matricielles telles que le collagène de type I, II, X, IX et XI [16-18] et la protéine de matrice oligomère du cartilage (COMP) [19] provoquent également d’autres formes de SMD. De plus, le phénotype moléculaire observé pour les mutations de collagène de type X ou COMP chevauche les mutations de la fibronectine, car elles provoquent également une rétention intracellulaire des protéines mutantes [20,21].
L’un des principaux objectifs de nos groupes de recherche est d’obtenir des informations pathomécanistes sur la façon dont les mutations de la fibronectine conduisent à la CF-SMD. Nous avons établi des cellules souches pluripotentes induites (iPSC) avec des fibroblastes cutanés donnés par des patients atteints de CF-SMD. Les iPSC sont un excellent outil pour modéliser et étudier les mécanismes physiologiques et pathologiques. Notre modèle de travail implique la différenciation des iPSCs en CSM, puis en chondrocytes, ce qui nous permet de définir les altérations des voies chondrogènes imposées par la fibronectine mutante. D’après nos travaux précédemment publiés [13,14], il est évident que ces mutations de la fibronectine causant une CF-SMD peuvent affecter les fonctions des cellules souches et leur différenciation par deux voies possibles. L’une est par accumulation intracellulaire de cette protéine matricielle qui peut conduire à un stress cellulaire, une autre est par carence en fibronectine dans le microenvironnement des cellules. Il a déjà été démontré qu’une matrice déficiente en fibronectine peut altérer la différenciation msc [8]. En outre, l’accumulation de protéines intracellulaires est également connue pour provoquer des dysplasies squelettiques, comme on l’a observé pour les mutations de SLC26A2 [22,23], de collagène de type X et de COMP [19]. Dans notre travail avec les iPSC de patients, nous analysons et évaluons comment les conséquences des mutations de la fibronectine pourraient se traduire en CF-SMD chez les personnes touchées.
À l’heure actuelle, seuls des traitements thérapeutiques très limités sont disponibles pour les patients atteints de dysplasie squelettique qui sont testés cliniquement et approuvés. La greffe de cellules souches, le traitement à l’hormone de croissance et l’utilisation de peptides synthétiques tels que les peptides natriurétiques de type C (achondroplasie) sont quelques-uns des traitements cliniques disponibles aujourd’hui [24]. Grâce à nos travaux de recherche en cours pour comprendre les mécanismes pathologiques des mutations de la fibronectine dans la CF-SMD, nous espérons avoir une meilleure idée de la façon dont la fibronectine dirige la différenciation des cellules souches en chondrocytes au niveau moléculaire et comment les mutations de la fibronectine modifient la fonction et la différenciation cellulaires. Nous espérons que nos résultats contribueront à l’avenir à identifier des cibles pharmacologiques dans le traitement de la CF-SMD et des troubles génétiques connexes.
À propos des auteurs
Dieter P. Reinhardt, Ph.D., est chercheur principal et Professeur distingué James McGill à la Faculté de médecine et des sciences de la santé et à la Faculté de médecine dentaire et des sciences de la santé buccodentaire de l’Université McGill. Ses recherches portent sur la détermination des mécanismes de la biogenèse et de la fonction des systèmes de fibres extracellulaires dans des conditions normales et pathologiques.
Le Dr Philippe Campeau, M.D., est professeur agrégé de clinique à l’Université de Montréal et chercheur principal au CHU Sainte-Justine. Ses travaux de recherche portent principalement sur l’étude des maladies épigénétiques, des dysplasies squelettiques, l’identification des gènes pathogènes et l’amélioration de la prise en charge des enfants touchés par ces affections.
Neha Dinesh, M.Sc. est étudiante au doctorat au Département d’anatomie et de biologie cellulaire de l’Université McGill et est co-supervisée par des professeurs Reinhardt et Campeau. Son projet de recherche consiste à étudier les conséquences des mutations de la fibronectine dans le développement du squelette et dans la dysplasie spondylométaphysaire.
Références
1. Tweedell KS. (2017) The adaptability of somatic stem cells: A review. J Stem Cells Regen Med 13, 3-13
2. Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, Lu WJ, Senarath-Yapa K, Chung MT, Marecic O, Tran M, Yan KS, Upton R, Walmsley GG, Lee AS, Sahoo D, Kuo CJ, Weissman IL, Longaker MT. (2015) Identification and specification of the mouse skeletal stem cell. Cell 160, 285-298
3. Matsushita Y, Ono W, Ono N. (2020) Skeletal stem cells for bone development and repair: Diversity matters. Curr Osteoporos Rep 18, 189-198
4. Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. (2008) Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 40, 46-62
5. Ono N, Ono W, Nagasawa T, Kronenberg HM. (2014) A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol 16, 1157-1167
6. Burton-Wurster N, Lust G, MacLeod JN. (1997) Cartilage fibronectin isoforms: In search of functions for a special population of matrix glycoproteins. Matrix Biol 15, 441-454
7. de Almeida PG, Pinheiro GG, Nunes AM, Goncalves AB, Thorsteinsdottir S. (2016) Fibronectin assembly during early embryo development: A versatile communication system between cells and tissues. Dev Dyn 245, 520-535
8. White ES, Muro AF. (2011) Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 63, 538-546
9. Zhang H, Chen X, Xue P, Ma X, Li J, Zhang J. (2021) FN1 promotes chondrocyte differentiation and collagen production via TGF-beta/PI3K/Akt pathway in mice with femoral fracture. Gene 769, 145-253
10. Casanova MR, Reis RL, Martins A, Neves NM. (2020) Fibronectin bound to a fibrous substrate has chondrogenic induction properties. Biomacromolecules 21, 1368-1378
11. Moursi AM, Damsky CH, Lull J, Zimmerman D, Doty SB, Aota S, Globus RK. (1996) Fibronectin regulates calvarial osteoblast differentiation. J Cell Sci 109, 1369-1380
12. Lee CS, Fu H, Baratang N, Rousseau J, Kumra H, Sutton VR, Niceta M, Ciolfi A, Yamamoto G, Bertola D, Marcelis CL, Lugtenberg D, Bartuli A, Kim C, Hoover-Fong J, Sobreira N, Pauli R, Bacino C, Krakow D, Parboosingh J, Yap P, Kariminejad A, McDonald MT, Aracena MI, Lausch E, Unger S, Superti-Furga A, Lu JT, Baylor-Hopkins Center for Mendelian Genomics, Cohn DH, Tartaglia M, Lee BH, *Reinhardt DP, *Campeau PM. *Co-senior authors (2017) Mutations in fibronectin cause a subtype of spondylometaphyseal dysplasia with “corner fractures”. Am J Hum Genet 101, 815-823
13. Cadoff EB, Sheffer R, Wientroub S, Ovadia D, Meiner V, Schwarzbauer JE. (2018) Mechanistic insights into the cellular effects of a novel FN1 variant associated with a spondylometaphyseal dysplasia. Clin Genet 94, 429-437
14. Costantini A, Valta H, Baratang NV, Yap P, Bertola DR, Yamamoto GL, Kim CA, Chen J, Wierenga KJ, Fanning EA, Escobar L, McWalter K, McLaughlin H, Willaert R, Begtrup A, Alm JJ, Reinhardt DP, Makitie O, Campeau PM. (2019) Novel fibronectin mutations and expansion of the phenotype in spondylometaphyseal dysplasia with « corner fractures ». Bone 121, 163-171
15. Marzin P, Cormier-Daire V. (2020) New perspectives on the treatment of skeletal dysplasia. Ther Adv Endocrinol Metab 11, 2042018820904016
16. Walter K, Tansek M, Tobias ES, Ikegawa S, Coucke P, Hyland J, Mortier G, Iwaya T, Nishimura G, Superti-Furga A, Unger S. (2007) COL2A1-related skeletal dysplasias with predominant metaphyseal involvement. Am J Med Genet 143A, 161-167
17. Krakow D. (2015) Skeletal dysplasias. Clin Perinatol 42, 301-319i
18. Gibson BG, Briggs MD. (2016) The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis 11, 86
19. Maddox BK, Keene DR, Sakai LY, Charbonneau NL, Morris NP, Ridgway CC, Boswell BA, Sussman MD, Horton WA, Bachinger HP, Hecht JT. (1997) The fate of cartilage oligomeric matrix protein is determined by the cell type in the case of a novel mutation in pseudoachondroplasia. J Biol Chem 272, 30993-30997
20. Hecht JT, Hayes E, Haynes R, Cole WG. (2005) COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol 23, 525-533
21. Wilson R, Freddi S, Chan D, Cheah KS, Bateman JF. (2005) Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J Biol Chem 280, 15544-15552
22. Galante LL, Schwarzbauer JE. (2007) Requirements for sulfate transport and the diastrophic dysplasia sulfate transporter in fibronectin matrix assembly. J Cell Biol 179, 999-1009
23. Zheng C, Lin X, Xu X, Wang C, Zhou J, Gao B, Fan J, Lu W, Hu Y, Jie Q, Luo Z, Yang L. (2019) Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias. EBioMedicine 40, 695-709
24. Jelin AC, O’Hare E, Blakemore K, Jelin EB, Valle D, Hoover-Fong J. (2017) Skeletal dysplasias: Growing therapy for growing bones. Front Pharmacol 8, 79
Crédit photo : Bright field image of a whole mount skeletal staining of a 1-day old mouse pup, avec l’aimable autorisation de Neha Dinesh. Mineralized bone (purple) is stained by alizarin red and cartilaginous skeleton (blue) is stained by alcian blue. L’os minéralisé (violet) est coloréé par le rouge alizarine et le squelette cartilagineux (bleu) est par le bleu alcian.