MRM Insights: Mesenchymal Stromal Cells and Fibrosis Modulation

Inés Colmegna

James G. Martin

Arina Morozan
Every month, in MRM Insights, a member of the MRM Network is writing about stem cells and regenerative medicine from a different perspective. This month, James G. Martin, Professor of Medicine at the RI-MUHC, Inés Colmegna, Associate Professor in the Division of Rheumatology at the RI-MUCH, and Arina Morozan, M.Sc. Candidate in their labs, are discussing Mesenchymal Stromal Cells and Fibrosis modulation.
Mesenchymal Stromal Cells and Fibrosis Modulation: Relevance of well characterized models
‘Pulmonary fibrosis’ comprises a group of heterogeneous lung diseases characterized by cellular proliferation, interstitial inflammation, fibrosis, or a combination of those findings within the alveolar wall1. In some cases, pulmonary fibrosis is associated with known causes (e.g. autoimmune diseases) and in others it is idiopathic2. A favored conceptual model of pulmonary fibrosis pathogenesis is that recurrent, subclinical epithelial injury superimposed on accelerated epithelial aging (i.e. senescence of alveolar epithelial cells and fibroblasts) that leads to aberrant repair of the injured alveolus and deposition of extracellular matrix by myofibroblasts1,3. A diagnosis of pulmonary fibrosis is associated with reduced survival and impaired quality of life with progressive hypoxemic respiratory failure4. Current pharmacological treatments fail to reverse fibrosis and only a fraction of pulmonary fibrosis patients qualify for lung transplantation5. The severity of pulmonary fibrosis and the lack of therapeutics to promote fibrosis resolution highlight the need for novel and effective treatments.
Animal models helped to identify pathways leading to lung fibrosis and to test the effect of preventive or therapeutic interventions. However, each model has inherent limitations and does not recapitulate every feature of pulmonary fibrosis. Common methods to induce fibrosis include systemic or intratracheal instillation of bleomycin, radiation, intratracheal administration of silica or asbestos, and transgenic mice or gene transfer employing fibrogenic mediators. Among those, the bleomycin model of lung fibrosis is still, by far the most common experimental tool in this field6. Bleomycin induces DNA strand breaks, resulting in inflammation, injury, and subsequent interstitial fibrosis. In addition to intratracheal instillation, there are multiple other ways to administer bleomycin, including intraperitoneal, subcutaneous and intravenous routes including using osmotic pumps7. Advantages of the osmotic minipump include that it allows better standardization of bleomycin delivery and causes fibrosis of the skin, lungs and other organs8. This multiorgan fibrotic involvement resembles what occurs in systemic fibrosing diseases such as scleroderma and allows evaluating the effect of anti-fibrotic agents in multiple organs. During 2021-22, we established the bleomycin osmotic minipump model and characterized it with pulmonary function tests, imaging modalities and histology. The reproducibility of these assays is encouraging and provides the opportunity to use them as readouts to test therapeutic agents in this model.
Multipotent mesenchymal stromal cells (MSCs) are progenitor cells that show intermediate levels of major histocompatibility complex (MHC) class I molecules on their cell surface and have no detectable levels of MHC class II, mainly HLA-DR, and costimulatory molecules (CD40, CD80, and CD86)9. This phenotype allows their allogeneic use for transplant. Despite the broad in vivo MSC distribution the most common sources of MSCs for therapeutic purposes are bone marrow (BM), adipose tissue (AT) and umbilical cord (UC). Growing evidence both from in vitro and clinical studies suggest that MSC’s properties vary according to their tissue of origin/source (i.e. ‘not all MSC are created equal’)10. Compared to BM-MSC, AT-MSC are reported to have higher proliferative capability, display a lower senescence rate, exhibit higher genetic stability, and have superior immunosuppressive properties11. This accounts for the increasing number of clinical trials using AT-MSC. On the other hand, compared to BM-MSC, UC-MSC show (1) richest stemness characteristics; (2) homogeneity (well-defined cell population with minimal contamination by other cell types); (3) larger numbers and rapid proliferation requiring minimal expansion, (4) broad differentiation potential and (5) hypo-immunogenicity12, 13. MSCs have immunomodulatory, pro-angiogenic, and less well-characterized anti-fibrotic properties14. Neither the presence of MSC’s nor their persistence in tissues is necessary for MSC clinical benefits15. This argues for a ‘hit and run’ mechanism mediated by paracrine factors (i.e. soluble molecules and extracellular vesicles) secreted by MSC’s16. In animals, after intravenous administration, most MSCs are trapped in the lungs and are rapidly removed from the circulation (half-life of ~ 24 hours) with persistence of their effects15.
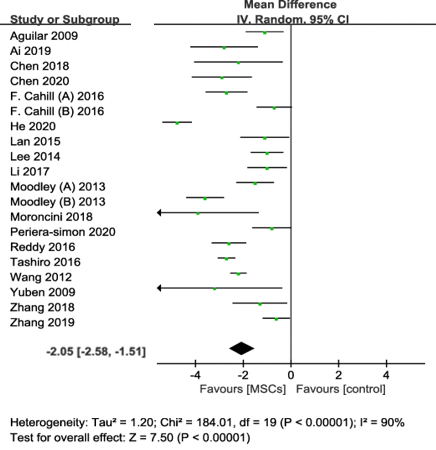
Figure 1 – Forest plot summarizing the effect of MSC therapy on pulmonary fibrosis scores
The ability of MSCs to regulate immunity, inhibit inflammation, and promote epithelial tissue repair highlight the promise of MSC therapy for treating pulmonary fibrosis. A recent meta-analysis of preclinical studies concluded that compared to the control group, MSC therapy was associated with an improvement in survival rate (odds ratios (OR) 3.10, 95% confidence interval (CI) 2.06 to 4.67, P < 0.001, I2 = 0%) and a significant reduction in pulmonary fibrosis scores (weighted mean difference 2.05, 95% CI −2.58 to −1.51, p < 0.001, I2 = 90%) (Figure 1 – modified from original publication17). These results provided the rationale for early human clinical trials that confirmed the safety of MSCs in the treatment of pulmonary fibrosis18. However, in those trials MSCs were associated with limited effects on clinical outcomes19. This strongly highlights the need for studies to inform the optimal MSC source and dose, route of administration, and frequency (single vs. multiple-dose regimens) for the treatment of pulmonary fibrosis. In addition, a better understanding of the anti-fibrotic mechanisms of MSC action, the development of in vitro assays to test the anti-fibrotic effects of MSCs and select based on the ‘best fitted MSC’s for this effect, knowing how the local microenvironment modulates MSC function, and developing strategies to potentiate the effects of MSCs are needed to improve the therapeutic effects of MSC in pulmonary fibrosis. This work requires collaboration integrating basic and clinical expertise. For this reason, McGill is in a unique position to move this agenda forward.
References
1. Lederer, D.J. & Martinez, F.J. Idiopathic Pulmonary Fibrosis. N Engl J Med 378, 1811-1823 (2018).
2. Hopkins, R.B., Burke, N., Fell, C., Dion, G. & Kolb, M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J 48, 187-195 (2016).
3. Alder, J.K. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A 105, 13051-13056 (2008).
4. Raghu, G. et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2, 566-572 (2014).
5. Kapnadak, S.G. & Raghu, G. Lung transplantation for interstitial lung disease. Eur Respir Rev 30 (2021).
6. Kolb, P. et al. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur Respir J 55 (2020).
7. Liu, T., De Los Santos, F.G. & Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol Biol 1627, 27-42 (2017).
8. Ravanetti, F. et al. Modeling pulmonary fibrosis through bleomycin delivered by osmotic minipump: a new histomorphometric method of evaluation. Am J Physiol Lung Cell Mol Physiol 318, L376-L385 (2020).
9. Le Blanc, K., Tammik, C., Rosendahl, K., Zetterberg, E. & Ringden, O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol 31, 890-896 (2003).
10. Galipeau, J. & Sensebe, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 22, 824-833 (2018).
11. Ribeiro, A. et al. Mesenchymal stem cells from umbilical cord matrix, adipose tissue and bone marrow exhibit different capability to suppress peripheral blood B, natural killer and T cells. Stem Cell Res Ther 4, 125 (2013).
12. Deuse, T. et al. Immunogenicity and immunomodulatory properties of umbilical cord lining mesenchymal stem cells. Cell Transplant 20, 655-667 (2011).
13. Davies, J.E., Walker, J.T. & Keating, A. Concise Review: Wharton’s Jelly: The Rich, but Enigmatic, Source of Mesenchymal Stromal Cells. Stem Cells Transl Med 6, 1620-1630 (2017).
14. Spees, J.L., Lee, R.H. & Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther 7, 125 (2016).
15. de Witte, S.F.H. et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells 36, 602-615 (2018).
16. Ferreira, J.R. et al. Mesenchymal Stromal Cell Secretome: Influencing Therapeutic Potential by Cellular Pre-conditioning. Front Immunol 9, 2837 (2018).
17. Yang, S. et al. Therapeutic Applications of Mesenchymal Stem Cells in Idiopathic Pulmonary Fibrosis. Front Cell Dev Biol 9, 639657 (2021).
18. Glassberg, M.K. et al. Allogeneic Human Mesenchymal Stem Cells in Patients With Idiopathic Pulmonary Fibrosis via Intravenous Delivery (AETHER): A Phase I Safety Clinical Trial. Chest 151, 971-981 (2017).
19. Cruz, F.F. & Rocco, P.R.M. The potential of mesenchymal stem cell therapy for chronic lung disease. Expert Rev Respir Med 14, 31-39 (2020).
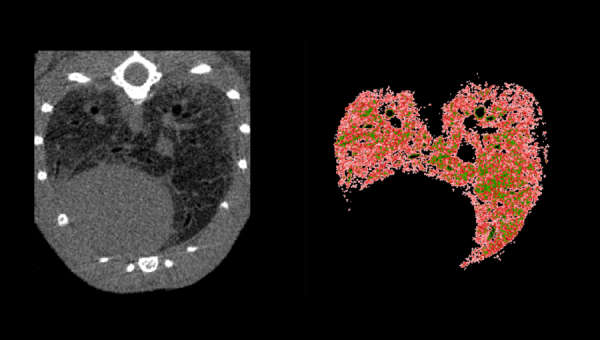
Image credit: Murin lung imaging by micro CT – Computerized tomographic image of murine lung following bleomycin administration by osmotic minipump over 10 days causing lung fibrosis (left) and a corresponding image illustrating poorly aerated lung (right) shown in red. Courtesy of Antonio Aliaga (image acquisition) & Guoming Xiong (image analysis).