MRM Insights : Cellules stromales mésenchymateuses et modulation de la fibrose

Inés Colmegna

James G. Martin

Arina Morozan
Chaque mois, dans les MRM Insights, un membre du Réseau MRM écrit sur les cellules souches et la médecine régénérative d’un point de vue différent. Ce mois-ci, James G. Martin, Professeur de médecine à l’IR-CUSM, Inés Colmegna, Professeure agrégée dans la Division de rhumatologie à l’IR-CUSM, et Arina Morozan, candidate à la maîtrise dans leurs laboratoires, discutent des cellules stromales mésenchymateuses dans la modulation de la fibrose.
Cellules stromales mésenchymateuses et modulation de la fibrose : Pertinence de modèles bien caractérisés
La « fibrose pulmonaire » comprend un groupe de maladies pulmonaires hétérogènes caractérisées par une prolifération cellulaire, une inflammation interstitielle, une fibrose ou une combinaison de ces résultats dans la paroi alvéolaire1. Dans certains cas, la fibrose pulmonaire est associée à des causes connues (par exemple, des maladies auto-immunes) et dans d’autres, elle est idiopathique2. Un modèle conceptuel privilégié de la pathogenèse de la fibrose pulmonaire est cette lésion épithéliale subclinique récurrente superposée au vieillissement épithélial accéléré (c’est-à-dire la sénescence des cellules épithéliales alvéolaires et des fibroblastes) qui conduit à une réparation aberrante de l’alvéole blessée et au dépôt de matrice extracellulaire par les myofibroblastes1,3. Un diagnostic de fibrose pulmonaire est associé à une survie réduite et à une altération de la qualité de vie avec une insuffisance respiratoire hypoxémique progressive4. Les traitements pharmacologiques actuels ne parviennent pas à inverser la fibrose et seule une fraction des patients atteints de fibrose pulmonaire sont admissibles à une transplantation pulmonaire5. La gravité de la fibrose pulmonaire et le manque de traitements pour favoriser la résolution de la fibrose soulignent la nécessité de traitements nouveaux et efficaces.
Les modèles animaux ont permis d’identifier les voies menant à la fibrose pulmonaire et de tester l’effet des interventions préventives ou thérapeutiques. Cependant, chaque modèle a des limites inhérentes et ne reproduit pas toutes les caractéristiques de la fibrose pulmonaire. Les méthodes courantes pour induire la fibrose comprennent l’instillation systémique ou intratrachéale de bléomycine, la radiothérapie, l’administration intratrachéale de silice ou d’amiante, et les souris transgéniques ou le transfert de gènes utilisant des médiateurs fibrogéniques. Parmi ceux-ci, le modèle de bléomycine de la fibrose pulmonaire est encore, de loin, l’outil expérimental le plus courant dans ce domaine6. La bléomycine induit des ruptures de brins d’ADN, entraînant une inflammation, des blessures et une fibrose interstitielle ultérieure. En plus de l’instillation intratrachéale, il existe de multiples autres façons d’administrer la bléomycine, y compris par voie intrapéritonéale, sous-cutanée et intraveineuse, ainsi qu’à l’aide de pompes osmotiques7. L’un des avantages de la minipompe osmotique est qu’elle permet une meilleure standardisation de l’administration de bléomycine et provoque une fibrose de la peau, des poumons et d’autres organes8. Cette atteinte fibrotique multiviscérale ressemble à ce qui se produit dans les maladies fibrosantes systémiques telles que la sclérodermie et permet d’évaluer l’effet des agents anti-fibrotiques dans plusieurs organes. En 2021-2022, nous avons établi le modèle de minipompe osmotique de bléomycine et l’avons caractérisé avec des tests de la fonction pulmonaire, des modalités d’imagerie et de l’histologie. La reproductibilité de ces tests est encourageante et offre la possibilité de les utiliser comme lectures pour tester des agents thérapeutiques dans ce modèle.
Les cellules stromales mésenchymateuses multipotentes (CSM) sont des cellules progénitrices qui présentent des niveaux intermédiaires de molécules majeures de classe I du complexe d’histocompatibilité (CMH) à la surface de leurs cellules et qui n’ont pas de niveaux détectables de CMH de classe II, principalement HLA-DR, ni de molécules costimulatrices (CD40, CD80 et CD86)9. Ce phénotype permet leur utilisation allogénique pour la transplantation. Malgré la large distribution in vivo des CSM, les sources les plus courantes de CSM à des fins thérapeutiques sont la moelle osseuse, le tissu adipeux et le cordon ombilical. De plus en plus de preuves provenant d’études in vitro et cliniques suggèrent que les propriétés des CSM varient en fonction de leur tissu d’origine/source (c’est-à-dire que « tous les CSM ne sont pas créées égales »)10. Par rapport aux CSM issues de la moelle épinière, les CSM du tissu adipeux auraient une capacité proliférative plus élevée, afficheraient un taux de sénescence plus faible, présenteraient une stabilité génétique plus élevée et auraint des propriétés immunosuppressives supérieures11. Cela explique le nombre croissant d’essais cliniques utilisant des CSM issues de tissu adipeux. D’autre part, par rapport aux CSM issues de la moelle épinière, les CSM de cordon ombilical présentent (1) des caractéristiques « souches » les plus riches; (2) une homogénéité (population cellulaire bien définie avec une contamination minimale par d’autres types de cellules); (3) un plus grand nombre et une prolifération rapide nécessitant une expansion minimale, (4) un large potentiel de différenciation et (5) une hypo-immunogénicité12, 13. Les CSM ont des propriétés immunomodulatrices, pro-angiogéniques et anti-fibrotiques moins bien caractérisées14. Ni la présence de CSM ni leur persistance dans les tissus ne sont nécessaires pour les avantages cliniques des CSM15. Cela plaide en faveur d’un mécanisme de « délit de fuite » médié par des facteurs paracrines (c’est-à-dire des molécules solubles et des vésicules extracellulaires) sécrétés par les CSM16. Chez les animaux, après administration intraveineuse, la plupart des CSM sont piégés dans les poumons et sont rapidement retirés de la circulation (demi-vie d’environ 24 heures) avec la persistance de leurs effets15.
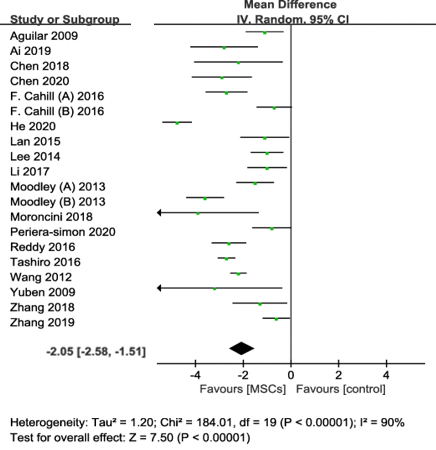
Figure 1 – Graphique en forêt résumant l’effet de la thérapie CSM sur les scores de fibrose pulmonaire.
La capacité des CSM à réguler l’immunité, à inhiber l’inflammation et à promouvoir la réparation des tissus épithéliaux souligne la promesse de la thérapie CSM pour le traitement de la fibrose pulmonaire. Une méta-analyse récente d’études précliniques a conclu que, par rapport au groupe témoin, le traitement par CSM était associé à une amélioration du taux de survie (rapports de cotes (RC) 3,10; intervalle de confiance (IC) à 95 % 2,06 à 4,67; P < 0,001, I2 = 0 %) et à une réduction significative des scores de fibrose pulmonaire (différence de moyenne pondérée de 2,05; IC à 95 % -2,58 à -1,51; p < 0,001; I2 = 90 %) (Figure 1 – modifiée à partir de la publication originale17). Ces résultats ont fourni la justification des premiers essais cliniques chez l’homme qui ont confirmé l’innocuité des CSM dans le traitement de la fibrose pulmonaire18. Cependant, dans ces essais, les CSM ont été associés à des effets limités sur les résultats cliniques19. Cela souligne fortement la nécessité d’études pour informer de la source et de la dose optimales de CSM, de la voie d’administration et de la fréquence (schémas posologiques à dose unique ou multiple) pour le traitement de la fibrose pulmonaire. En outre, une meilleure compréhension des mécanismes anti-fibrotiques de l’action des CSM, le développement de tests in vitro pour tester les effets anti-fibrotiques des CSM et sélectionner en fonction les CSM les mieux adaptées à cet effet, en sachant comment le microenvironnement local module la fonction du CSM et en développant des stratégies pour potentialiser les effets des CSM sont nécessaires pour améliorer les effets thérapeutiques des CSM dans la fibrose pulmonaire. Ce travail nécessite une collaboration intégrant l’expertise de base et clinique. Pour cette raison, McGill est dans une position unique pour faire avancer ce programme.
Références
1. Lederer, D.J. & Martinez, F.J. Idiopathic Pulmonary Fibrosis. N Engl J Med 378, 1811-1823 (2018).
2. Hopkins, R.B., Burke, N., Fell, C., Dion, G. & Kolb, M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J 48, 187-195 (2016).
3. Alder, J.K. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A 105, 13051-13056 (2008).
4. Raghu, G. et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2, 566-572 (2014).
5. Kapnadak, S.G. & Raghu, G. Lung transplantation for interstitial lung disease. Eur Respir Rev 30 (2021).
6. Kolb, P. et al. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur Respir J 55 (2020).
7. Liu, T., De Los Santos, F.G. & Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol Biol 1627, 27-42 (2017).
8. Ravanetti, F. et al. Modeling pulmonary fibrosis through bleomycin delivered by osmotic minipump: a new histomorphometric method of evaluation. Am J Physiol Lung Cell Mol Physiol 318, L376-L385 (2020).
9. Le Blanc, K., Tammik, C., Rosendahl, K., Zetterberg, E. & Ringden, O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol 31, 890-896 (2003).
10. Galipeau, J. & Sensebe, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 22, 824-833 (2018).
11. Ribeiro, A. et al. Mesenchymal stem cells from umbilical cord matrix, adipose tissue and bone marrow exhibit different capability to suppress peripheral blood B, natural killer and T cells. Stem Cell Res Ther 4, 125 (2013).
12. Deuse, T. et al. Immunogenicity and immunomodulatory properties of umbilical cord lining mesenchymal stem cells. Cell Transplant 20, 655-667 (2011).
13. Davies, J.E., Walker, J.T. & Keating, A. Concise Review: Wharton’s Jelly: The Rich, but Enigmatic, Source of Mesenchymal Stromal Cells. Stem Cells Transl Med 6, 1620-1630 (2017).
14. Spees, J.L., Lee, R.H. & Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther 7, 125 (2016).
15. de Witte, S.F.H. et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells 36, 602-615 (2018).
16. Ferreira, J.R. et al. Mesenchymal Stromal Cell Secretome: Influencing Therapeutic Potential by Cellular Pre-conditioning. Front Immunol 9, 2837 (2018).
17. Yang, S. et al. Therapeutic Applications of Mesenchymal Stem Cells in Idiopathic Pulmonary Fibrosis. Front Cell Dev Biol 9, 639657 (2021).
18. Glassberg, M.K. et al. Allogeneic Human Mesenchymal Stem Cells in Patients With Idiopathic Pulmonary Fibrosis via Intravenous Delivery (AETHER): A Phase I Safety Clinical Trial. Chest 151, 971-981 (2017).
19. Cruz, F.F. & Rocco, P.R.M. The potential of mesenchymal stem cell therapy for chronic lung disease. Expert Rev Respir Med 14, 31-39 (2020).
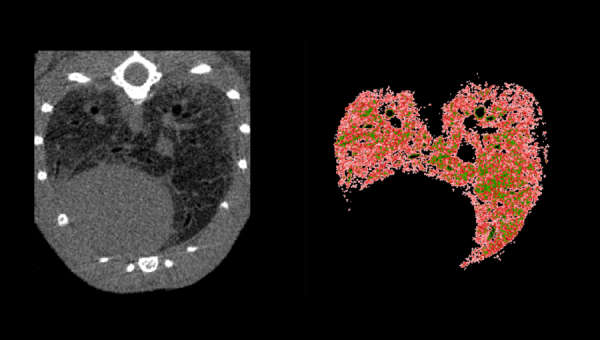
Crédits de l’image : Murin lung imaging by micro CT – Image tomographique informatisée du poumon murin après administration de bléomycine par minipompe osmotique sur 10 jours provoquant une fibrose pulmonaire (à gauche) et image correspondante illustrant un poumon mal aéré (à droite) en rouge. Avec l’aimable autorisation d’Antonio Aliaga (acquisition d’images) et Guoming Xiong (analyse d’images).