MRM Insights: Measuring Protein Turnover in IPSC-Derived Brain Organoids

Jerry Dong

Jean-François Trempe
Every month, in MRM Insights, a member of the MRM Network is writing about stem cells and regenerative medicine from a different perspective. This month, Jean-François Trempe, Associate Professor in the Department of Pharmacology & Therapeutics, and Jerry Dong, MSc student in his lab, are discussing how to measure protein turnover in iPSC-derived brain organoids.
Measuring Protein Turnover in IPSC-Derived Brain Organoids
If a ship remained in a harbor and a single wooden plank was replaced one at a time until all its original pieces were replaced, would it still fundamentally be the same ship? Analogous to the Ship of Theseus, the human body and its organs are constantly undergoing turnover as small parts and their constituent cellular pieces are being replaced over time. Almost all of your cells are completely replaced after a decade thus rendering you to be an entirely new entity since your conception. Yet, it is only when the process of removing decaying cellular proteins is harmoniously replaced with the synthesis of new functional proteins, do we retain our sense of an immutable self. Indeed, the body’s ability to recycle damaged proteins is essential to maintaining homeostasis and living a healthy life. If these proteins are not removed, they can accumulate leading to the production of misfolded proteins and gain toxic properties that further harm the cell [1]. One example of this demonstrates that failure in the proteasome, a protein complex that is essential to recycling components of the cell, is associated with neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s Disease [2]. Different studies have also shown that autophagy, another cellular pathway involved in protein and organelle degradation, is a key regulator of aggregate-prone proteins such as alpha-synuclein, TDP-43, and tau proteins [3]. Mutant forms of these proteins are also classical markers of neurodegenerative diseases. Hence, developing drugs to modulate these degradative pathways to restore the clearance of these harmful components is an exciting prospect for the treatment of these diseases.
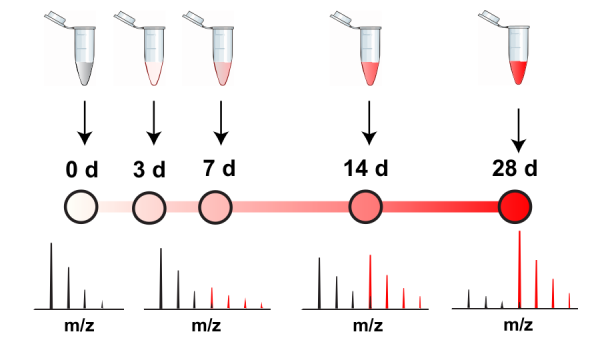
However, before we can develop therapeutics to regulate these degradation pathways, we must first be able to identify how these pathways are initially compromised in a diseased state. This requires us to measure protein turnover in healthy cells so a baseline can be established when comparing turnover rates with defective cells. How does one then measure protein dynamics in cells? Turnover can be measured using stable isotope–labelled amino acids (SILAC) [4]. These labelled amino acids “tracers” are heavier than their normal constituents in proteins and can be detected using mass spectrometry following their incorporation in newly synthesized proteins. The experiment starts with cells or animals that are first fed with normal (“light”) amino acids. The cells or animals are then “pulsed” or exposed to nutrients enriched with the “heavy” amino acids. We can then measure the rate at which the tracer is being incorporated into specific proteins to measure its half-life.
The following Figure 1 is a simplified schematic that illustrates this concept. Imagine a cell with free-floating peptides that belong to proteins X and Y. Over the course of 28 days, the cells will be exposed to an excess of the SILAC tracer, a labelled form of the amino acid leucine in this case. Protein X and Y will incorporate the tracer over time and we can measure what percentage of the peptides have incorporated the tracer using mass spectrometry. We can see that protein Y is turned over faster than protein X as it reaches a higher tracer percentage at each time point. From the tracer percentages, we can also compute a half-life for each given protein.
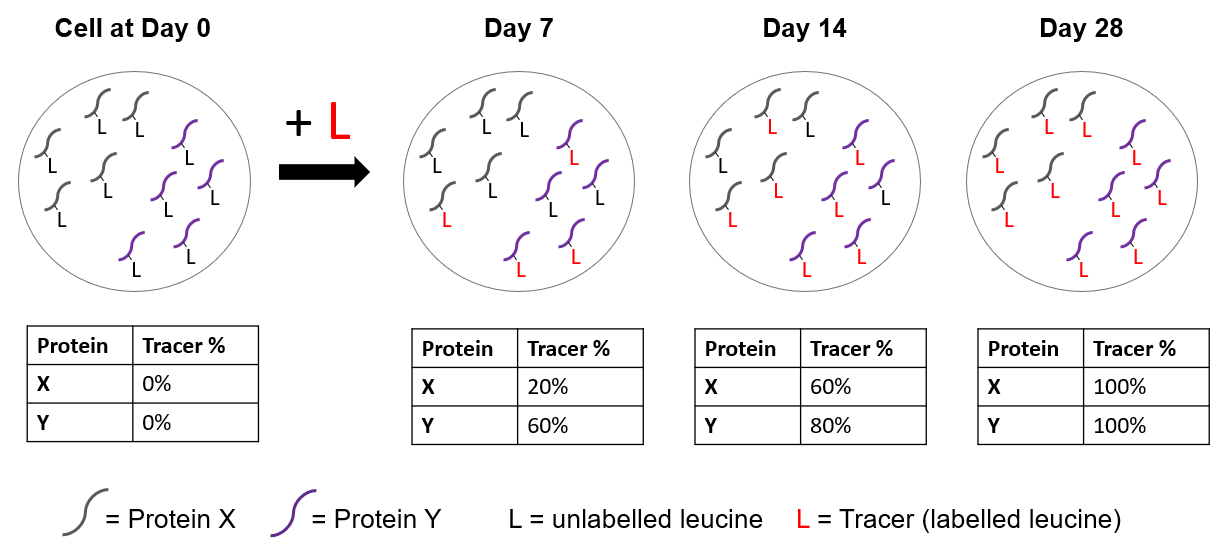
Figure 1: Schematic demonstrating two theoretical proteins with different half-lives incorporating the labelled tracer at separate time points.
There are a few points of caution that accompany the example mentioned above. Firstly, we are functioning under the assumption that the cells or animals are in a steady state throughout the course of the labeling experiment (ie. the organism is not continuously growing). At steady state, the rate of protein synthesis is equal to the rate of protein degradation and thus the global protein levels remain unchanged. Secondly, when a tracer is first introduced to a cell, it is diluted by unlabelled amino acids found as free amino acids or generated by the breakdown of pre-existing proteins [5]. Hence, the amino acid pool contributing to newly synthesized protein contains a mixture of both unlabelled and labelled amino acids that must be accounted for. For more information on how to handle these precautions, we and others have published protocols to measure the turnover of proteins from such labelling experiments with mass spectrometry data [5-7].
How can these methods be translated to actual tangible discoveries or treatments that benefit patients? Through intersecting these techniques used to measure protein turnover with the disease modeling capabilities of stem cells, we can shed light onto underlying mechanisms of disorders that have previously eluded the scientific community. For example, organoids, three-dimensional self-organizing culture systems derived from human-induced Pluripotent Stem Cells (iPSCs), have emerged as a promising tool for capturing diseases [8]. Their ability to recapitulate the architecture and complexity of a human organ with remarkable detail extends beyond conventional two-dimensional in vitro cell cultures and animal models. For instance, amyloid-β plaques and neurofibrillary tangles are pathological hallmarks of AD that are not fully captured in human neurons derived from patients with AD [9]. Human neural organoids with familial AD mutations serve to bridge that gap as studies have demonstrated the successful recapitulation of key features such as amyloid-β and tau pathology in these 3D culture systems [9]. Likewise, midbrain-like organoids (MBOs) reproduce key features of substantia nigra dopaminergic neurons, which are particularly vulnerable in PD [10]. Our colleagues Vi-Nguyen Mohamed et al. here at McGill indeed showed that MBOs with a familial PD mutation display aggregation of the protein α-synuclein, a hallmark feature of PD [11].
In order to identify degradation pathways that are affected in diseases, it is also essential that protein half-lives in organoid models are comparable to those observed in mammalian brains. Thus, our team developed a robust workflow to measure protein turnover in human MBOs using a SILAC-based time course approach [6]. We compared the half-lives of 773 proteins in MBOs derived from wild-type or a knockout of Parkin, an E3 ubiquitin ligase involved in mitochondrial quality control. Using a similar SILAC time course, it was previously observed that fruit flies with a deletion of Parkin exhibit slower turnover in a subset of mitochondrial proteins implicated in respiration [12]. Surprisingly, we found no major differences in the turnover of mitochondrial proteins between wild-type and Parkin-KO hMBOs. However, a group of proteins typically found in astrocytes had a faster turnover in Parkin-KO, suggesting that these non-neuronal cells were activated, consistent with previous observations in human PD patients. Furthermore, we were also able to compare our protein half-lives with previously published studies and found that hMBOs half-lives correlate better with those measured in mouse brain cortex than with primary cortical neurons. In future work, we intend to analyze hMBOs with different familial PD mutations to determine whether they perturb protein turnover in shared pathways, which would provide a unifying basis to understand why some neurons are more affected than others in PD.
Thus, measurement of protein turnover in organoids has the potential to unravel the mystery behind neurodegenerative diseases. However, significant challenges remain to make the methods more widely applicable and powerful. While 773 proteins may seem like a lot, it represents less than 5% of the entire human proteome. Furthermore, it is currently not possible to determine the cell of origin of a given identified protein. Recent advances in the sensitivity and speed of mass spectrometers coupled with single-cell technologies will enable us to explore in much greater depth the proteome dynamics of individual cells in the nervous system. This will lead to the identification of new degradation pathways and proteins that can be targeted for the development of therapeutics.
References
1. Fecto, F., Esengul, Y.T. & Siddique, T. (2014). Protein recycling pathways in neurodegenerative diseases. Alz Res Therapy 6, 13. https://doi.org/10.1186/alzrt243
2. Zheng, Q., Huang, T., Zhang, L., Zhou, Y., Luo, H., Xu, H., & Wang, X. (2016). Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Frontiers in aging neuroscience, 8, 303. https://doi.org/10.3389/fnagi.2016.00303
3. Frake, R. A., Ricketts, T., Menzies, F. M., & Rubinsztein, D. C. (2015). Autophagy and neurodegeneration. The Journal of clinical investigation, 125(1), 65–74. https://doi.org/10.1172/JCI73944
4. Ross, A. B., Langer, J. D., & Jovanovic, M. (2021). Proteome Turnover in the Spotlight: Approaches, Applications, and Perspectives. Molecular & cellular proteomics : MCP, 20, 100016. https://doi.org/10.1074/mcp.R120.002190
5. Hsieh, E. J., Shulman, N. J., Dai, D. F., Vincow, E. S., Karunadharma, P. P., Pallanck, L., Rabinovitch, P. S., & MacCoss, M. J. (2012). Topograph, a software platform for precursor enrichment corrected global protein turnover measurements. Molecular & cellular proteomics : MCP, 11(11), 1468–1474. https://doi.org/10.1074/mcp.O112.017699
6. Dong, J., Duchesne, A., Bayne, A. N., Mohamed, N. V., Yi, W., Mathur, M., Chen, C., You, Z., Abdian, N., Taylor, L., Fon, E. A., Durcan, T. M., & Trempe, J. F. (2022). An approach to measuring protein turnover in human induced pluripotent stem cell organoids by mass spectrometry. Methods (San Diego, Calif.), 203, 17–27. https://doi.org/10.1016/j.ymeth.2022.03.011
7. Price, J. C., Guan, S., Burlingame, A., Prusiner, S. B., & Ghaemmaghami, S. (2010). Analysis of proteome dynamics in the mouse brain. Proceedings of the National Academy of Sciences of the United States of America, 107(32), 14508–14513. https://doi.org/10.1073/pnas.1006551107
8. Kim, J., Koo, B. K., & Knoblich, J. A. (2020). Human organoids: model systems for human biology and medicine. Nature reviews. Molecular cell biology, 21(10), 571–584. https://doi.org/10.1038/s41580-020-0259-3
9. Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’Avanzo, C., Chen, H., Hooli, B., Asselin, C., Muffat, J., Klee, J. B., Zhang, C., Wainger, B. J., Peitz, M., Kovacs, D. M., Woolf, C. J., Wagner, S. L., Tanzi, R. E., & Kim, D. Y. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature, 515(7526), 274–278. https://doi.org/10.1038/nature13800
10. Jo, J.et al. (2016). Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell stem cell, 19(2), 248–257. https://doi.org/10.1016/j.stem.2016.07.005
11. Mohamed, N. V. et al. (2021). Midbrain organoids with an SNCA gene triplication model key features of synucleinopathy. Brain communications, 3(4), fcab223. https://doi.org/10.1093/braincomms/fcab223
12. Vincow, E. S., Merrihew, G., Thomas, R. E., Shulman, N. J., Beyer, R. P., MacCoss, M. J., & Pallanck, L. J. (2013). The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proceedings of the National Academy of Sciences of the United States of America, 110(16), 6400–6405. https://doi.org/10.1073/pnas.1221132110