MRM Insights: Modeling ALS in 3D with iPSC-derived cells

María José Castellanos Montiel

Mathile Chaineau

Thomas Durcan
In the MRM Insights, a member of the MRM Network offers a unique perspective on the topics of stem cells and regenerative medicine. In this edition, Dr. Thomas Durcan, who serves as an Associate Professor at The Neuro and Director of the Early Drug Discovery Unit (EDDU), collaborates with Mathilde Chaineau, Program Manager and Research Associate at the EDDU, and graduate student María José Castellanos Montiel. Together, they discuss how to model ALS in 3D with iPSC-derived cells.
Modeling ALS in 3D with iPSC-derived cells
Amyotrophic Lateral Sclerosis (ALS) is a fatal disease affecting upper and lower motor neurons (MNs) resulting in weakness and atrophy of the skeletal muscles. The lack of models recapitulating the disease in humans as well as the heterogeneity of the disease between patients make the development of therapies challenging and despite decades of research, ALS currently has no cure and only limited options of treatment. Mechanisms underlying MNs degeneration remain unclear. To date mechanisms and pathways involved in excitotoxicity, oxidative stress, disruption of the endolysosomal pathway and autophagy or dysregulation of protein homeostasis and RNA processing are known to be involved in the development and progression of the disease, but the heterogeneity of the disease makes it difficult to identify and prioritize targets for ALS patients. If intrinsic properties of populations of MNs make them vulnerable (cell-autonomous component), additional cell types also play a role in the disease (non-cell autonomous component), including but not restricted to astrocytes and microglia.
Astrocytes are glial cells from the central nervous system (CNS) where one of their functions is to promote neuronal survival by secreting trophic factors beneficial to neurons, and by regulating neurotransmitter levels in the extracellular space, or excess of glutamate for instance, through its recapture by the glutamate transporter EAAT2. However, in the context of ALS and other neurodegenerative diseases, and through mechanisms that remain not well elucidated, astrocytes can turn on the neurons by becoming reactive and promoting neuronal damages that can lead to their loss, and/or by losing their homeostatic maintenance function. For example, astrocytes with various mutations in SOD1, C9orf72 genes or astrocytes derived from patient with sporadic ALS affect the survival of mice motor neurons (1, 2) or human MNs (3, 4).
Microglia are the resident immune cells of the CNS and undergo morphological changes and respond rapidly through an induction of genetic programs designed to overcome and repair CNS insults when they detect injuries, degeneration, or infection. Microglia have become central to many studies on neurodegenerative diseases. With respect to ALS in particular, studies investigating disease progression in ALS mice indicate that, in vivo, resident microglia increase their number during disease progression, and their activation states represent a continuum between the two classical phenotypes i.e., neuroprotective vs. neuroactive (5). Activated microglia isolated from ALS mice models overexpressing mSOD1 exhibit neuroprotective features at the onset of the disease as opposed to the cells isolated from mice at more advanced stage of the disease which exhibits neurotoxic effects when co-cultured with MNs. Moreover, adult SOD1-G93A microglia significantly decrease survival of mice MNs within a few days through the NF-κB pathway (6).
Neuroinflammation is traditionally studied using glial cells conditioned media or by co-culturing MNs with astrocytes or microglia and overall, the interplay between microglia and astrocytes in ALS development and progression is understudied with only a few studies published both in mice and post-mortem tissues (7, 8). At The Neuro’s Early Drug Discovery Unit (EDDU), through a project funded by ALS Canada and Brain Canada, and in collaboration with Dr. Iturria-Medina’s lab from the Neuroimaging and Neuroinformatics Research Group at The Neuro, we aim to better understand the non-cell autonomous component of disease progression at the cellular level, using induced pluripotent stem cells (iPSCs) derived from patients with ALS. Such iPSCs can be readily differentiated into any cell type under the appropriate conditions (9, 10), providing a platform to study the different cells present within the human brain. For our project, to model ALS in a dish, we propose using iPSCs from patients with familial or sporadic form of ALS that we will direct into human spinal MNs, spinal cord-like astrocytes and microglia to be co-cultured together in a 3D triculture spheroid and to study their interactions. This human model of ALS in a dish combined with computational analysis normally applied to clinical and imaging data from large cohorts of patients to model the development of neurological diseases will enable us to study the progression of the disease to acquire longitudinal imaging, survival, and functional data.
The disassembling of neuromuscular junctions (NMJs), the connections between motor nerve terminals and muscle fibers is another key element in the onset and progression of ALS. Since the late sixties, researchers have tried to create models of the neuromuscular junction (NMJ) in a dish (11), aiming to study the mechanisms affecting this structure in a subset of diseases called neuromuscular disorders, which include ALS. Historically, the evolution of these models has been driven by the goal of moving each subsequent model closer to mimicking the real-life conditions of the neuromuscular junction. In this context, the next generation of NMJ models in a dish considers three key aspects: 1) growing the cells in a three-dimensional fashion like they are found in the body (12-14); 2) physically distancing the MNs from the muscle cells using special devices in order to replicate the spatial gap between the spinal cord, where the MNs reside, and the various muscles through the body (14, 15); and 3) building models that include a greater amount of cellular types that directly or indirectly influence NMJ function (15, 16).
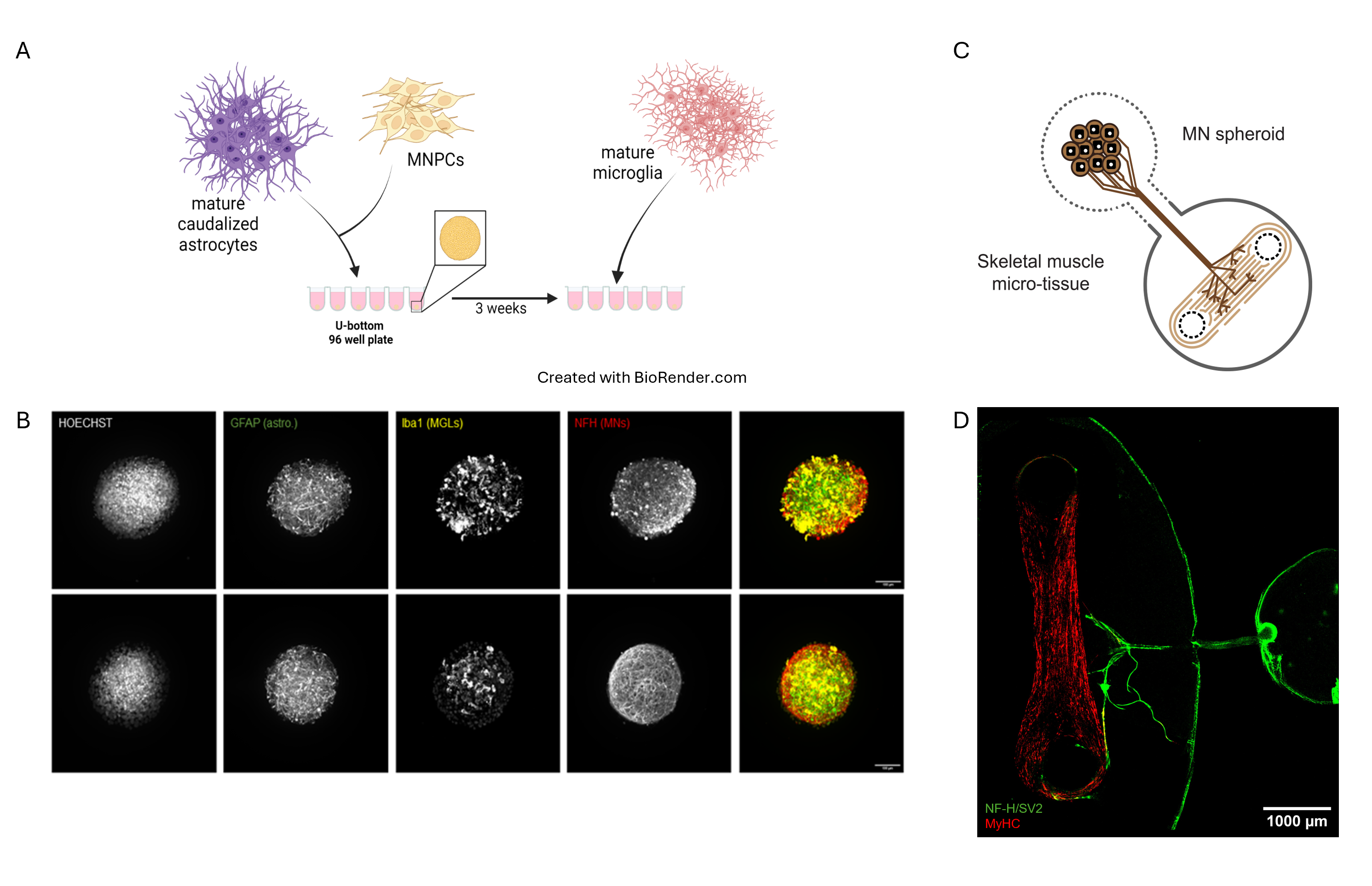
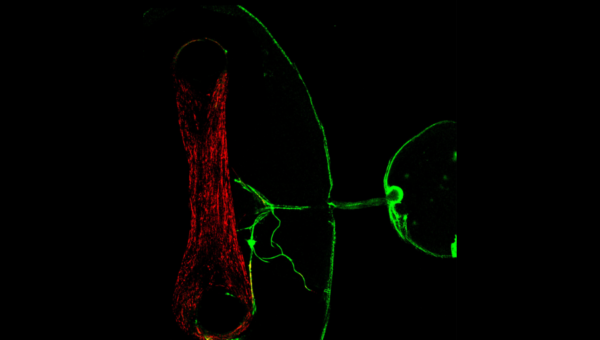
3D models of ALS. (A) Schematic of the generation of the 3D triculture with MNs, astrocytes and microglia. (B) A spheroid was first generated with iPSC derived MNs and mature caudalized astrocytes. After 3 weeks of co-culture, microglia was added onto the spheroid and the triculture spheroid was maturated for an additional week (top panel) or for 3 weeks (bottom panel). (C) Schematic of the human 3D NMJ model inside a microfluidic device allowing axons of the MNs to connect with the skeletal muscle. (D) After 21 to 28 days of co-culture, the MN spheroid has grown axons (NF-H/SV2 staining) that contact the muscle (MyHC staining) in the skeletal muscle micro-tissue chamber.
At the EDDU, we successfully optimized a protocol to cultivate iPSC-derived MNs and human skeletal muscle in a three-dimensional fashion within a dish. A key component of this protocol is the use of a microfluidic device that enables us to establish a spatial separation between MNs and skeletal muscle, closely mirroring body conditions. Furthermore, we established different methodologies to visualize and analyze the morphology of our NMJs, as well as workflows towards assessing their functional characteristics. As a follow-up of the ALS Canada – Brain Canada Discovery grant, our group is interested in going one step further by incorporating the 3D spheroids tri-culture into our NMJ model and studying how the interactions between MNs, astrocytes and microglia lead to NMJ impairment. Collectively, growing the cells in a three-dimensional environment, recreating the spatial organization of the body, and adding other cellular types brings us closer in building a more physiological representation of the NMJs that can help to recapitulate the features of ALS in a dish, helping to increase our understanding into why it develops and progresses as it does.
References
1. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824-8.
2. Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;111(2):829-32.
3. Birger A, Ben-Dor I, Ottolenghi M, Turetsky T, Gil Y, Sweetat S, et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine. 2019;50:274-89.
4. Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3(6):637-48.
5. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237(1):147-52.
6. Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81(5):1009-23.
7. Jha MK, Jo M, Kim JH, Suk K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist. 2019;25(3):227-40.
8. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481-7.
9. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861-72.
10. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-76.
11. Castellanos-Montiel MJ, Velasco I, Escobedo-Avila I. Modeling the neuromuscular junction in vitro: an approach to study neuromuscular junction disorders. Ann N Y Acad Sci. 2021;1488(1):3-15.
12. Afshar Bakooshli M, Lippmann ES, Mulcahy B, Iyer N, Nguyen CT, Tung K, et al. A 3D culture model of innervated human skeletal muscle enables studies of the adult neuromuscular junction. Elife. 2019;8.
13. Faustino Martins JM, Fischer C, Urzi A, Vidal R, Kunz S, Ruffault PL, et al. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell. 2020;26(2):172-86.e6.
14. Osaki T, Uzel SGM, Kamm RD. On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat Protoc. 2020;15(2):421-49.
15. Machado CB, Pluchon P, Harley P, Rigby M, Gonzalez Sabater V, Stevenson DC, et al. In Vitro Modelling of Nerve-Muscle Connectivity in a Compartmentalised Tissue Culture Device. Adv Biosyst. 2019;3(7).
16. Pereira JD, DuBreuil DM, Devlin AC, Held A, Sapir Y, Berezovski E, et al. Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat Commun. 2021;12(1):4744.