MRM Insights : Modélisation de la SLA en 3D avec des cellules dérivées d’iPSC

María José Castellanos Montiel

Mathile Chaineau

Thomas Durcan
Dans les MRM Insights, les membres du Réseau de médecine régénérative de McGill offrent une perspective unique sur les sujets des cellules souches et de la médecine régénérative. Dans cette édition, le Dr Thomas Durcan, qui est Professeur Associé au Neuro et Directeur de l’Unité de Découverte Précoce de Médicaments (PDMPP), collabore avec Mathilde Chaineau, Gestionnaire de Programme et Associée de Recherche à la PDMPP, ainsi qu’avec l’étudiante María José Castellanos Montiel. Ensemble, ils discutent de la modélisation de la SLA en 3D avec des cellules dérivées d’iPSC.
Modélisation de la SLA en 3D avec des cellules dérivées d’iPSC
La sclérose latérale amyotrophique (SLA) est une maladie mortelle qui affecte les neurones moteurs supérieurs et inférieurs, entraînant une faiblesse et une atrophie des muscles squelettiques. Le manque de modèles reproduisant la maladie chez l’homme ainsi que l’hétérogénéité de la maladie entre les patients rendent le développement de thérapies difficile, et malgré des décennies de recherche, la SLA n’a actuellement aucun remède et seulement des options de traitement limitées. Les mécanismes sous-jacents à la dégénérescence des neurones moteurs restent peu clairs. À ce jour, les mécanismes et les voies impliqués dans l’excitotoxicité, le stress oxydatif, la perturbation de la voie endolysosomale et de l’autophagie, ou la dysrégulation de l’homéostasie des protéines et du traitement de l’ARN sont connus pour être impliqués dans le développement et la progression de la maladie, mais l’hétérogénéité de la maladie rend difficile l’identification et la priorisation des cibles pour les patients atteints de SLA. Si les propriétés intrinsèques des populations de neurones moteurs les rendent vulnérables (composante cellulaire autonome), d’autres types de cellules jouent également un rôle dans la maladie (composante non cellulaire autonome), notamment mais sans s’y limiter, les astrocytes et les microglies.
Les astrocytes sont des cellules gliales du système nerveux central (SNC) dont l’une des fonctions est de favoriser la survie neuronale en sécrétant des facteurs trophiques bénéfiques pour les neurones, et en régulant les niveaux de neurotransmetteurs dans l’espace extracellulaire, ou l’excès de glutamate par exemple, en le recapturant via le transporteur de glutamate EAAT2. Cependant, dans le contexte de la SLA et d’autres maladies neurodégénératives, et par des mécanismes qui restent mal élucidés, les astrocytes peuvent devenir réactifs et favoriser les dommages neuronaux pouvant conduire à leur perte, et/ou perdre leur fonction de maintien de l’homéostasie. Par exemple, les astrocytes porteurs de diverses mutations dans les gènes SOD1, C9orf72 ou dérivés de patients atteints de SLA sporadique affectent la survie des neurones moteurs chez les souris (1, 2) ou les neurones moteurs humains (3, 4).
Les microglies sont les cellules immunitaires résidentes du système nerveux central, subissant des changements morphologiques et qui répondent rapidement par l’induction de programmes génétiques conçus pour surmonter et réparer les dommages du SNC lorsqu’elles détectent des blessures, une dégénérescence ou une infection. Les microglies sont devenues centrales dans de nombreuses études sur les maladies neurodégénératives. En ce qui concerne la SLA en particulier, des études sur la progression de la maladie chez les souris atteintes de SLA indiquent qu’in vivo, les microglies résidentes augmentent leur nombre au cours de la progression de la maladie, et leurs états d’activation représentent un continuum entre les deux phénotypes classiques, à savoir neuroprotecteur vs. neuroactif. Les microglies activées isolées à partir de modèles murins de SLA surexprimant mSOD1 présentent des caractéristiques neuroprotectrices au début de la maladie par opposition aux cellules isolées chez les souris à un stade plus avancé de la maladie qui présentent des effets neurotoxiques lorsqu’elles sont en co-culture avec des neurones moteurs. De plus, les microglies adultes SOD1-G93A diminuent significativement la survie des neurones moteurs chez les souris en quelques jours via la voie NF-κB.
La neuroinflammation est traditionnellement étudiée en utilisant le milieu conditionné des cellules gliales ou en co-cultivant des neurones moteurs avec des astrocytes ou des microglies, et dans l’ensemble, l’interaction entre les microglies et les astrocytes dans le développement et la progression de la SLA est peu étudiée, avec seulement quelques études publiées sur les souris et les tissus post-mortem. Au sein de la Plateforme de découverte de médicaments en phase précoce (PDMPP) du Neuro, dans le cadre d’un projet financé par ALS Canada et Brain Canada, et en collaboration avec le laboratoire du Dr Iturria-Medina du groupe de recherche en neuroimagerie et neuroinformatique du Neuro, nous visons à mieux comprendre la composante non-autonome de la progression de la maladie au niveau cellulaire, en utilisant des cellules souches pluripotentes induites (iPSCs) dérivées de patients atteints de SLA. De telles iPSCs peuvent être facilement différenciées en tout type de cellule dans des conditions appropriées, offrant une plateforme pour étudier les différentes cellules présentes dans le cerveau humain. Pour notre projet, afin de modéliser la SLA en culture, nous proposons d’utiliser des iPSCs de patients atteints de forme familiale ou sporadique de SLA que nous différencierons en neurones moteurs spinaux humains, en astrocytes semblables à la moelle épinière et en microglies, afin de les co-cultiver ensemble dans une tri-culture sphéroïde en 3D et d’étudier leurs interactions. Ce modèle humain de SLA en culture, combiné à une analyse computationnelle généralement appliquée aux données cliniques et d’imagerie provenant de vastes cohortes de patients pour modéliser le développement des maladies neurologiques, nous permettra d’étudier la progression de la maladie pour acquérir des données d’imagerie longitudinales, de survie et fonctionnelles.
Le désassemblage des jonctions neuromusculaires (JNM), les connexions entre les terminaisons nerveuses motrices et les fibres musculaires, est un autre élément clé dans le déclenchement et la progression de la SLA. Depuis la fin des années soixante, les chercheurs ont tenté de créer des modèles de jonctions neuromusculaires en culture, dans le but d’étudier les mécanismes affectant cette structure dans un sous-ensemble de maladies appelées troubles neuromusculaires, qui incluent la SLA. Historiquement, l’évolution de ces modèles a été guidée par l’objectif de rapprocher chaque modèle subséquent de la reproduction des conditions réelles de la jonction neuromusculaire. Dans ce contexte, la prochaine génération de modèles de JNM en culture prend en compte trois aspects clés : 1) faire croître les cellules de manière tridimensionnelle comme elles se trouvent dans le corps ; 2) éloigner physiquement les neurones moteurs des cellules musculaires en utilisant des dispositifs spéciaux afin de reproduire l’écart spatial entre la moelle épinière, où résident les neurones moteurs, et les différents muscles à travers le corps ; et 3) construire des modèles qui incluent une plus grande quantité de types cellulaires qui influencent directement ou indirectement la fonction des JNMs.
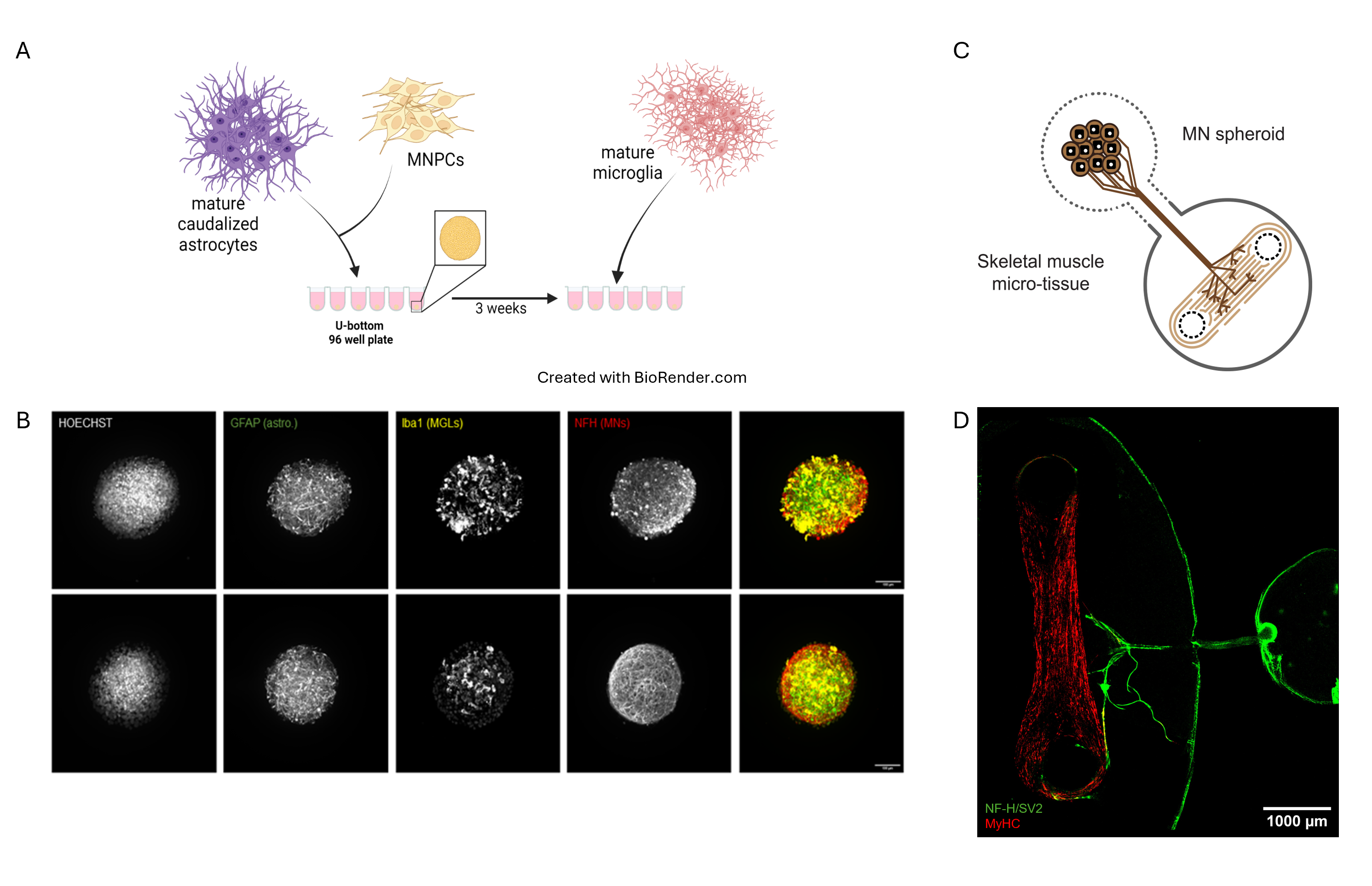
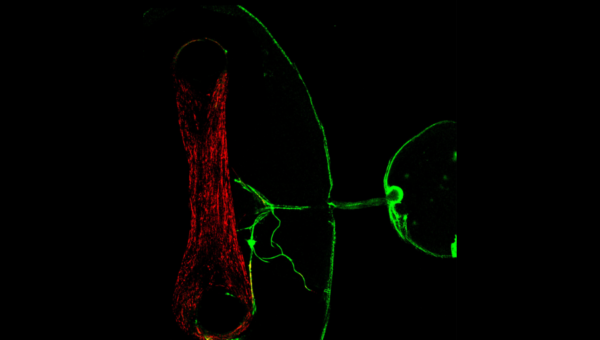
Modèles 3D de la SLA. (A) Schéma de la génération de la triculture 3D avec des MNs, des astrocytes et des microglies. (B) Une sphéroïde a d’abord été générée avec des MNs dérivés d’iPSC et des astrocytes caudalisés matures. Après 3 semaines de co-culture, les microglies ont été ajoutées sur la sphéroïde et la sphéroïde triculture a été maturée pendant une semaine supplémentaire (panneau supérieur) ou pendant 3 semaines (panneau inférieur). (C) Schéma du modèle JNM humain 3D à l’intérieur d’un dispositif microfluidique permettant aux axones des MNs de se connecter avec le muscle squelettique. (D) Après 21 à 28 jours de co-culture, la sphéroïde de MN a développé des axones (marquage NF-H/SV2) qui contactent le muscle (marquage MyHC) dans la chambre de micro-tissu musculaire squelettique.
À la PDMPP, nous avons réussi à optimiser un protocole pour cultiver des neurones moteurs dérivés d’iPSCs et des muscles squelettiques humains de manière tridimensionnelle dans une culture. Un élément clé de ce protocole est l’utilisation d’un dispositif microfluidique qui nous permet d’établir une séparation spatiale entre les neurones moteurs et les muscles squelettiques, reproduisant étroitement les conditions du corps. De plus, nous avons mis en place différentes méthodologies pour visualiser et analyser la morphologie de nos JNMs, ainsi que des flux de travail pour évaluer leurs caractéristiques fonctionnelles. Dans le cadre du suivi de la subvention Discovery d’ALS Canada – Brain Canada, notre groupe est intéressé à aller encore plus loin en incorporant la triculture en sphéroïdes 3D dans notre modèle de JNM et à étudier comment les interactions entre les neurones moteurs, les astrocytes et les microglies conduisent à l’altération des JNMs. Dans l’ensemble, la croissance des cellules dans un environnement tridimensionnel, la recréation de l’organisation spatiale du corps et l’ajout d’autres types cellulaires nous rapprochent de la construction d’une représentation plus physiologique des JNMs qui peut aider à reproduire les caractéristiques de la SLA dans une culture, contribuant ainsi à augmenter notre compréhension de la raison pour laquelle elle se développe et progresse comme elle le fait.
Références
1. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824-8.
2. Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;111(2):829-32.
3. Birger A, Ben-Dor I, Ottolenghi M, Turetsky T, Gil Y, Sweetat S, et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine. 2019;50:274-89.
4. Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3(6):637-48.
5. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237(1):147-52.
6. Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81(5):1009-23.
7. Jha MK, Jo M, Kim JH, Suk K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist. 2019;25(3):227-40.
8. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481-7.
9. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861-72.
10. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-76.
11. Castellanos-Montiel MJ, Velasco I, Escobedo-Avila I. Modeling the neuromuscular junction in vitro: an approach to study neuromuscular junction disorders. Ann N Y Acad Sci. 2021;1488(1):3-15.
12. Afshar Bakooshli M, Lippmann ES, Mulcahy B, Iyer N, Nguyen CT, Tung K, et al. A 3D culture model of innervated human skeletal muscle enables studies of the adult neuromuscular junction. Elife. 2019;8.
13. Faustino Martins JM, Fischer C, Urzi A, Vidal R, Kunz S, Ruffault PL, et al. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell. 2020;26(2):172-86.e6.
14. Osaki T, Uzel SGM, Kamm RD. On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat Protoc. 2020;15(2):421-49.
15. Machado CB, Pluchon P, Harley P, Rigby M, Gonzalez Sabater V, Stevenson DC, et al. In Vitro Modelling of Nerve-Muscle Connectivity in a Compartmentalised Tissue Culture Device. Adv Biosyst. 2019;3(7).
16. Pereira JD, DuBreuil DM, Devlin AC, Held A, Sapir Y, Berezovski E, et al. Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat Commun. 2021;12(1):4744.