MRM Insights : l’autophagie dans les cellules souches saines et malades

Cordell VanGenderen

Dre Natasha Chang
Chaque mois, dans les MRM Insights, un membre du Réseau MRM écrit sur les cellules souches et la médecine régénérative d’un point de vue différent. Ce mois-ci, la Dre Natasha Chang, Professeure adjointe au Département de biochimie à l’Université McGill, et Cordell VanGenderen, étudiant au doctorat dans son laboratoire, discutent de l’autophagie dans les cellules souches saines et malades.
L’autophagie dans les cellules souches saines et malades
L’autophagie (qui signifie littéralement auto-alimentation) est le processus par lequel les cellules décomposent leurs composants internes pour recycler les biomolécules constitutives. Chaque cellule a un niveau basal d’autophagie, qui peut être élevé en réponse à des stimuli tels que la privation de nutriments et l’accumulation d’espèces réactives de l’oxygène (reactive oxygen species, ROS). Des recherches antérieures sur les cellules différenciées en phase terminale ont révélé un rôle essentiel pour l’autophagie dans le maintien de la santé cellulaire par l’élimination des organites et des protéines endommagés, ainsi que dans l’apport de nutriments et d’énergie en période de stress métabolique. L’autophagie elle-même fait référence à trois mécanismes distincts: la macroautophagie, la microautophagie et l’autophagie à médiation chaperonne. La macroautophagie est la plus étudiée et la mieux comprise de ces trois, et aux fins de cet article sera appelée autophagie. En bref, cette forme spécifique d’autophagie implique la formation d’une structure à double membrane (connue sous le nom de phagophore) qui enveloppe la cargaison à dégrader (Figure 1). L’achèvement du phagophore génère une vésicule connue sous le nom d’autophagosome, qui fusionne ensuite avec un lysosome, formant l’autolysosome. Les enzymes du lysosome décomposent ensuite le contenu de l’autolysosome et les composants constitutifs sont recyclés. Cette forme d’autophagie peut être sélective, comme on le voit dans le cas de la mitophagie, où les mitochondries sont spécifiquement ciblées par l’autophagosome pour la dégradation (Parzych & Klionsky, 2014). Des défauts dans l’autophagie ont été observés dans une variété de maladies, y compris les troubles neurodégénératifs, les maladies cardiaques et les myopathies (Wirawan et coll., 2011).
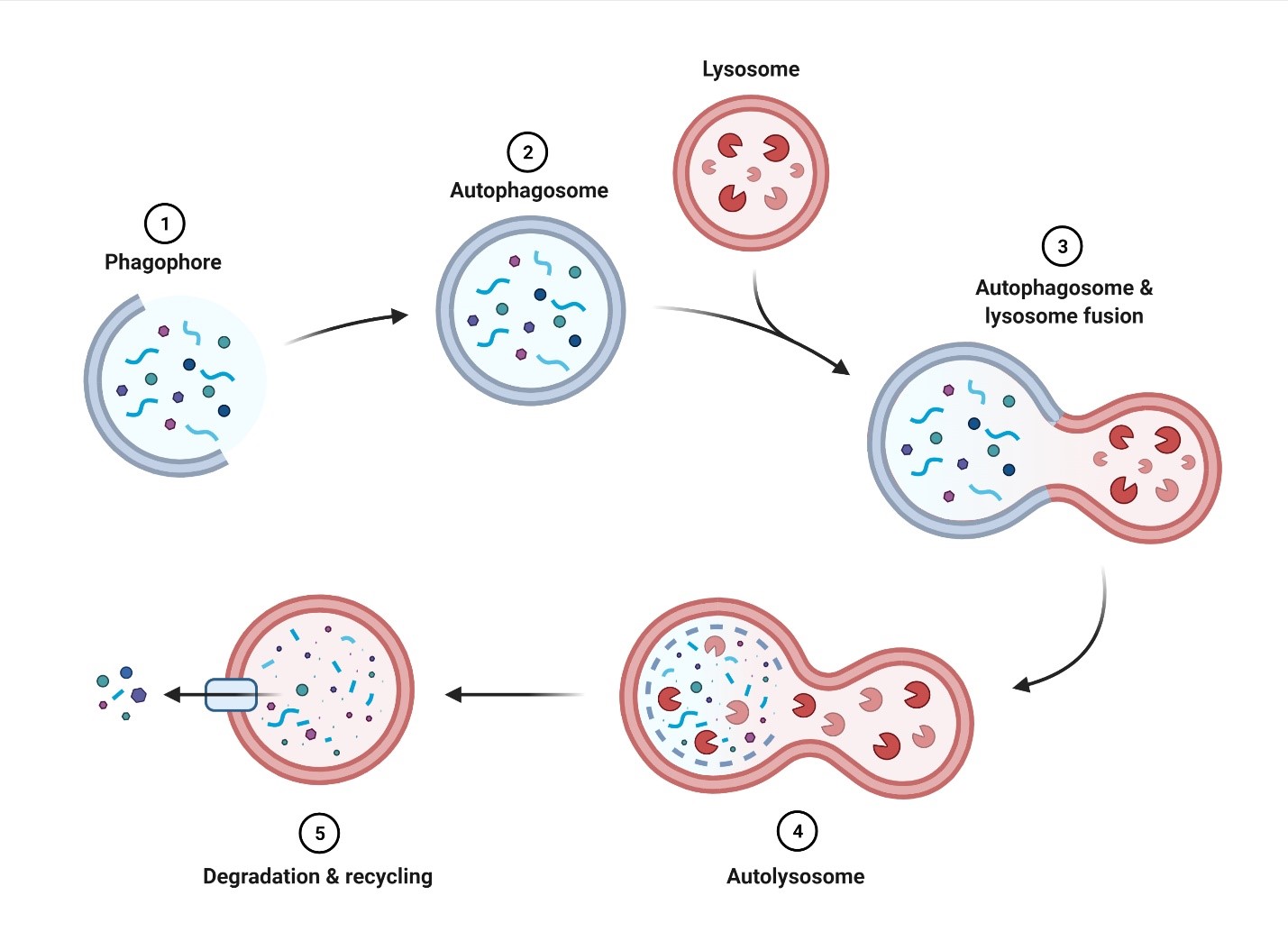
Figure 1. Vue d’ensemble de l’autophagie. 1) Allongement du phagophore entourant la cargaison à dégrader. 2) L’enceinte complète de la cargaison entraîne la formation de l’autophagosome. 3) Fusion de l’autophagosome et du lysosome. 4) Formation de l’autolysosome lors de la fusion complète de l’autophagosome et du lysosome. 5) La cargaison dans l’autolysosome est dégradée pour être réutilisée par la cellule. Figure réalisée à l’aide de BioRender.com.
Au cours des dernières années, on a de plus en plus apprécié le rôle que joue l’autophagie dans la biologie et la fonction des cellules souches multipotentes, et les implications potentielles de cette relation pour la maladie au-delà du maintien général de la santé cellulaire. Les cellules souches multipotentes sont définies comme celles qui conservent la capacité d’auto-renouvellement des cellules souches pluripotentes, mais qui sont limitées dans leur capacité à se différencier, étant confinées à une – ou quelques – lignées (Mirzaei et coll., 2018). En 2016, García-Prat et collaborateurs ont découvert que l’autophagie est un processus vital pour le maintien du caractère souche des cellules souches musculaires, également connues sous le nom de cellules satellites (García-Prat et coll., 2016). Pendant l’homéostasie, les cellules satellites (comme c’est typique avec les cellules souches multipotentes) restent en quiescence, un état réversible d’arrêt du cycle cellulaire. Lors de la stimulation (par exemple une blessure musculaire), ils s’activent et s’engagent dans la différenciation myogénique. Il a été découvert qu’en inhibant directement l’autophagie par la délétion spécifique dans les cellules satellites du gène clé de l’autophagie Atg7, les cellules satellites sont sorties de leur état de repos et sont entrées en sénescence. La sénescence est un état irréversible d’arrêt du cycle cellulaire, rendant ainsi les cellules satellites incapables de s’activer, de proliférer et de participer à la réparation musculaire. La perte d’autophagie induit une élévation du stress oxydatif résultant de l’accumulation de ROS, conséquence du cumul de mitochondries endommagées. Dans les cellules satellites, il en résulte l’expression de p16INK4, qui inhibe la kinase 4 dépendante des cyclines pour induire l’arrêt du cycle cellulaire et la sénescence ultérieure. Dans des cellules satellites âgées, ils ont pu inverser l’état de sénéscence grâce à un traitement à la rapamycine, un inhibiteur de la cible mammalienne du complexe de rapamycine I (mTORC1) et inducteur de l’autophagie. L’autophagie sert également à soutenir d’autres fonctions clés dans les cellules souches multipotentes qui sont importantes pour leur capacité à se différencier. Tout d’abord, l’autophagie est nécessaire pour répondre aux demandes énergétiques nécessaires aux cellules souches lorsqu’elles subissent une activation (Tang & Rando, 2014). Deuxièmement, l’autophagie est cruciale pour le remodelage cellulaire qui se produit lorsque les cellules souches subissent une différenciation, ce qui a été observé dans les cellules progénitrices cardiaques, les cellules souches hématopoïétiques et les cellules germinales (Huang et coll., 2020; Joshi et Kundu, 2013; Lampert et coll., 2019).
Le lien entre l’autophagie et les cellules souches est évident dans les maladies liées à l’âge. Continuons la discussion sur le dysfonctionnement des cellules souches musculaires. La sarcopénie est une perte de muscle squelettique et de leur fonction liée à l’âge. Lors de l’examen de la fonction des cellules satellites chez des souris âgées (20-24 mois) par rapport à des souris gériatriques (28-32 mois), Sousa-Victor et collaborateurs ont effectué une expérience de lésion musculaire et de régénération pour étudier l’activation et la réparation musculaire ultérieures des cellules satellites. L’altération de la capacité de régénération des souris gériatriques n’était pas due au nombre de cellules satellites, car les souris âgées et gériatriques présentaient un nombre similaire de cellules satellites. Cependant, les souris gériatriques ont présenté une plus grande prévalence de cellules satellites en sénescence, démontrant que l’incapacité d’activer les cellules satellites conduit à une régénération musculaire inefficace qui correspond à la sarcopénie (Sousa-Victor et coll., 2014). Couplé avec les résultats de García-Prat et collaborateurs, qui ont révélé que l’autophagie est importante pour prévenir la sénescence dans les cellules satellites, il est clair que le déclin lié à l’âge de l’autophagie des cellules satellites entraîne une altération de la régénération musculaire et aboutit à une perte musculaire.
Une autre maladie liée à l’âge directement touchée par l’autophagie des cellules souches est l’ostéoporose. En 2020, Yuan et collaborateurs ont démontré un lien entre la perte d’autophagie dans les cellules souches hématopoïétiques (hematopoietic stem cell, HSC) et l’ostéoporose (Yuan et coll., 2020). Ils ont observé une diminution du nombre de globules rouges des patients atteints d’ostéoporose, indiquant une perturbation de la différenciation et de la prolifération des HSC. Après examen d’échantillons de moelle osseuse de ces patients, ils ont constaté une réduction significative de l’expression des gènes autophagiques. Une expérience de suivi impliquant la délétion génétique d’Atg7 dans les HSC de souris (mais pas les lignées différenciées en phase terminale) a entraîné une perte osseuse sévère et une homéostasie osseuse perturbée, indiquant ainsi un lien entre l’autophagie des HSC et l’ostéoporose.
Dans la dystrophie musculaire de Duchenne (DMD), une maladie dégénérative musculaire que nous étudions dans le laboratoire Chang, l’autophagie s’est avérée altérée dans les cellules musculaires matures de souris mdx, un modèle murin de DMD (Palma et coll., 2012). Cependant, à l’époque, le rôle potentiel des cellules satellites dans la progression de la DMD n’était pas bien compris. En 2015, Dumont et collaborateurs ont découvert que les cellules satellites mdx étaient intrinsèquement altérées et que le dysfonctionnement des cellules satellites est un mécanisme contribuant à la DMD (Dumont et coll., 2015). Les cellules satellites dystrophiques présentent une division asymétrique altérée des cellules souches et favorisent l’auto-renouvellement par rapport à l’engagement, ce qui entraîne une réduction du nombre de cellules progénitrices. Bien que la contribution de l’autophagie des cellules souches dans le contexte de la DMD n’ait pas été clairement établie, nous émettons l’hypothèse que l’autophagie altérée joue un rôle dans le dysfonctionnement des cellules satellites dans la DMD, ce qui représente un objectif de recherche clé dans notre laboratoire.
L’hyperactivité des voies autophagiques dans les cellules souches multipotentes a également été liée à la maladie. Dans le foie résident des cellules stellaires hépatiques, qui restent au repos jusqu’à leur activation en réponse à une lésion hépatique, un processus qui repose sur l’autophagie (Thoen et coll., 2011). Lors de leur activation, les cellules stellaires commencent à se différencier en myofibroblastes, qui sont la principale source de collagène dans la matrice extracellulaire du foie (MEC). L’accumulation de collagène dans la MEC entraîne une fibrose hépatique contribuant à de multiples maladies chroniques du foie (Higashi et coll., 2017). Au cours de la fibrose hépatique, plusieurs voies de signalisation sont dérégulées d’une manière qui favorise l’autophagie, y compris la transformation du facteur de croissance β, HMGB1 (high-mobility group box-1) et la protéine IGFBP-rP1 (insulin-like growth factor-binding protein-related protein-1). Par conséquent, la suractivation de la signalisation pro-autophagie dans les cellules stellaires entraîne une activation et une différenciation inappropriées, contribuant à une maladie hépatique chronique (Li et coll., 2018; Zhang et coll., 2021; Zhou et coll., 2020).
La recherche au cours de ces dernières années a établi une tendance claire, selon laquelle la perturbation de l’autophagie dans les cellules souches adultes contribue à une myriade de maladies. Cette vision mécaniste a ouvert la porte à des opportunités thérapeutiques. Il existe de nombreux agents pharmacologiques capables de moduler l’autophagie, directement ou indirectement. La rapamycine, qui est la plus connue (ainsi que la pléthore de mimétiques de rapamycine appelés rapalogues) agit par inhibition de mTOR (Blagosklonny, 2019). mTOR est un régulateur central de la croissance cellulaire, servant de sous-unité kinase des complexes mTORC1 et mTORC2.
En réponse à l’abondance des nutriments, ces complexes protéiques s’activent et phosphorylent une multitude de protéines pour induire la croissance cellulaire, y compris les acteurs critiques de l’autophagie ULK1 et le facteur de transcription EB (TFEB). La phosphorylation de l’ULK1 et du TFEB par mTORC1 inhibe leur activité et empêche l’autophagie. ULK1 est responsable de la formation du complexe d’initiation ULK1 qui induit la formation de phagophores, tandis que TFEB induit la biogenèse des lysosomes (Jung et coll., 2010; Raben et Puertollano, 2016). Une autre méthode d’induire l’autophagie consiste à activer la protéine kinase activée par l’adénosine monophosphate (AMPK), qui phosphoryle et régule négativement mTOR tout en régulant positivement ULK1 et TFEB (Jung et coll., 2010). L’AMPK peut être activée par la metformine, un médicament contre le diabète de type 2 qui inhibe le complexe I de la chaîne respiratoire mitochondriale pour augmenter la quantité d’AMP, stimulant ainsi l’activité de l’AMPK (Kim et coll., 2016). Il est important de noter que mTOR et AMPK sont deux protéines régulatrices métaboliques qui ont un impact sur un large éventail de protéines pour induire des réponses au stress et l’autophagie (AMPK) ou les inhiber (mTOR). Par conséquent, le ciblage de ces kinases métaboliques peut entraîner des conséquences indésirables. Par exemple, la rapamycine est un modulateur immunitaire bien documenté et est utilisée pour prévenir le rejet d’organes (Blagosklonny, 2019). Une autre approche consiste à cibler directement la machinerie d’autophagie elle-même, dans laquelle près de 100 protéines sont spécifiquement impliquées. Une cible potentielle est le complexe d’initiation ULK1. L’agent pharmacologique LYN-1604 a été identifié comme un agoniste du complexe ULK et, lorsqu’il a été testé dans un essai sur le cancer du sein triple négatif, il s’est avéré qu’il induisait la mort cellulaire liée à l’autophagie (Xiang et coll., 2020). Bien qu’il soit peu probable qu’un seul agent soit en mesure de traiter toutes les maladies discutées ici, des études fondamentales visant à découvrir le rôle de l’autophagie des cellules souches dans la maladie fourniront des informations mécanistes importantes et identifieront de nouvelles cibles qui modulent l’autophagie pour les approches de médecine régénérative.
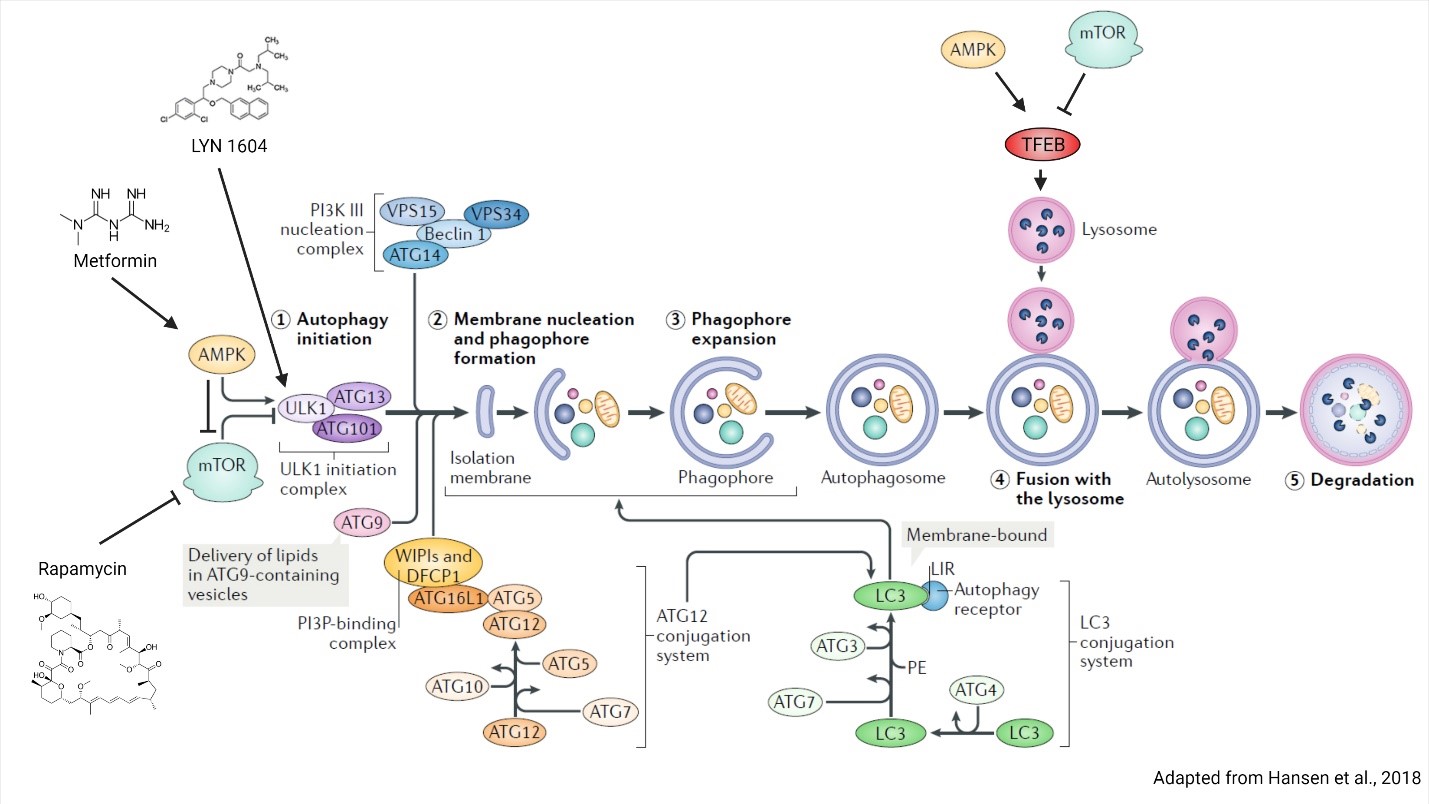
Figure 2. Régulation de l’autophagie par AMPK et mTOR. L’AMPK et mTOR régulent directement l’autophagie en régulant la biogenèse des phagophores et des lysosomes. La formation de phagophores est régulée par phosphorylation du complexe ULK1, tandis que la biogenèse des lysosomes est contrôlée par l’activité transcriptionnelle du TFEB. Les composés induisant l’autophagie, comme la rapamycine, la metformine et LYN 1604, et leurs cibles protéiques sont indiqués. La figure est adaptée de Hansen et coll., 2018.
Références
Blagosklonny, M. V. (2019). Rapamycin for longevity: Opinion article. Aging, 11(19), 8048–8067. https://doi.org/10.18632/aging.102355
Dumont, N. A., Wang, Y. X., Von Maltzahn, J., Pasut, A., Bentzinger, C. F., Brun, C. E., & Rudnicki, M. A. (2015). Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nature Medicine, 21(12), 1455–1463. https://doi.org/10.1038/nm.3990
García-Prat, L., Martínez-Vicente, M., Perdiguero, E., Ortet, L., Rodríguez-Ubreva, J., Rebollo, E., Ruiz-Bonilla, V., Gutarra, S., Ballestar, E., Serrano, A. L., Sandri, M., & Muñoz-Cánoves, P. (2016). Autophagy maintains stemness by preventing senescence. Nature, 529(7584), 37–42. https://doi.org/10.1038/nature16187
Hansen, M., Rubinsztein, D. C., & Walker, D. W. (2018). Autophagy as a promoter of longevity: insights from model organisms. Nature Reviews Molecular Cell Biology, 19(9), 579–593. https://doi.org/10.1038/s41580-018-0033-y
Higashi, T., Friedman, S. L., & Hoshida, Y. (2017). Hepatic stellate cells as key target in liver fibrosis. Advanced Drug Delivery Reviews, 121, 27–42. https://doi.org/10.1016/J.ADDR.2017.05.007
Huang, Q., Liu, Y., Zhang, S., Yap, Y. T., Li, W., Zhang, D., Gardner, A., Zhang, L., Song, S., Hess, R. A., & Zhang, Z. (2020). Autophagy core protein ATG5 is required for elongating spermatid development, sperm individualization and normal fertility in male mice. Autophagy, 1–15. https://doi.org/10.1080/15548627.2020.1783822/SUPPL_FILE/KAUP_A_1783822_SM7639.ZIP
Joshi, A., & Kundu, M. (2013). Mitophagy in hematopoietic stem cells: The case for exploration. In Autophagy (Vol. 9, Issue 11, pp. 1737–1749). https://doi.org/10.4161/auto.26681
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., & Kim, D. H. (2010). MTOR regulation of autophagy. In FEBS Letters (Vol. 584, Issue 7, pp. 1287–1295). https://doi.org/10.1016/j.febslet.2010.01.017
Kim, J., Yang, G., Kim, Y., Kim, J., & Ha, J. (2016). AMPK activators: mechanisms of action and physiological activities. Experimental & Molecular Medicine, 48(4), e224. https://doi.org/10.1038/EMM.2016.16
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., Kim, T., Sussman, M. A., & Gustafsson, Å. B. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation) BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor c. Autophagy, 15(7), 1182–1198. https://doi.org/10.1080/15548627.2019.1580095
Li, J., Zeng, C., Zheng, B., Liu, C., Tang, M., Jiang, Y., Chang, Y., Song, W., Wang, Y., & Yang, C. (2018). HMGB1-induced autophagy facilitates hepatic stellate cells activation: A new pathway in liver fibrosis. Clinical Science, 132(15), 1645–1667. https://doi.org/10.1042/CS20180177
Mirzaei, H., Sahebkar, A., Sichani, L. S., Moridikia, A., Nazari, S., Nahand, J. S., Salehi, H., Stenvang, J., Masoudifar, A., Mirzaei, H. R., & Jaafari, M. R. (2018). Therapeutic application of multipotent stem cells. Journal of Cellular Physiology, 233(4), 2815–2823. https://doi.org/10.1002/JCP.25990/FORMAT/PDF
Palma, C. De, Morisi, F., Cheli, S., Pambianco, S., Cappello, V., Vezzoli, M., Rovere-Querini, P., Moggio, M., Ripolone, M., Francolini, M., Sandri, M., & Clementi, E. (2012). Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death & Disease, 3(11), e418. https://doi.org/10.1038/CDDIS.2012.159
Parzych, K. R., & Klionsky, D. J. (2014). An overview of autophagy: Morphology, mechanism, and regulation. In Antioxidants and Redox Signaling (Vol. 20, Issue 3, pp. 460–473). Mary Ann Liebert, Inc. https://doi.org/10.1089/ars.2013.5371
Raben, N., & Puertollano, R. (2016). TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. In Annual Review of Cell and Developmental Biology (Vol. 32, pp. 255–278). Annual Reviews. https://doi.org/10.1146/annurev-cellbio-111315-125407
Sousa-Victor, P., Gutarra, S., García-Prat, L., Rodriguez-Ubreva, J., Ortet, L., Ruiz-Bonilla, V., Jardí, M., Ballestar, E., González, S., Serrano, A. L., Perdiguero, E., & Muñoz-Cánoves, P. (2014). Geriatric muscle stem cells switch reversible quiescence into senescence. Nature, 506(7488), 316–321. https://doi.org/10.1038/nature13013
Tang, A. H., & Rando, T. A. (2014). Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. The EMBO Journal, 33(23), 2782–2797. https://doi.org/10.15252/embj.201488278
Thoen, L. F. R., Guimarães, E. L. M., Dollé, L., Mannaerts, I., Najimi, M., Sokal, E., & Van Grunsven, L. A. (2011). A role for autophagy during hepatic stellate cell activation. Journal of Hepatology, 55(6), 1353–1360. https://doi.org/10.1016/j.jhep.2011.07.010
Wirawan, E., Vanden Berghe, T., Lippens, S., Agostinis, P., & Vandenabeele, P. (2011). Autophagy: for better or for worse. Cell Research, 22, 43–61. https://doi.org/10.1038/cr.2011.152
Xiang, H., Zhang, J., Lin, C., Zhang, L., Liu, B., & Ouyang, L. (2020). Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharmaceutica Sinica B, 10(4), 569–581. https://doi.org/10.1016/J.APSB.2019.10.003
Yuan, Y., Fang, Y., Zhu, L., Gu, Y., Li, L., Qian, J., Zhao, R., Zhang, P., Li, J., Zhang, H., Yuan, N., Zhang, S., Ma, Q., Wang, J., & Xu, Y. (2020). Deterioration of hematopoietic autophagy is linked to osteoporosis. Aging Cell, 19(5). https://doi.org/10.1111/acel.13114
Zhang, J., Jiang, N., Ping, J., & Xu, L. (2021). TGF-β1-induced autophagy activates hepatic stellate cells via the ERK and JNK signaling pathways. International Journal of Molecular Medicine, 47(1), 256–266. https://doi.org/10.3892/ijmm.2020.4778
Zhou, Y., Zhang, Q., Kong, Y., Guo, X., Zhang, H., Fan, H., & Liu, L. (2020). Insulin-Like Growth Factor Binding Protein-Related Protein 1 Activates Primary Hepatic Stellate Cells via Autophagy Regulated by the PI3K/Akt/mTOR Signaling Pathway. Digestive Diseases and Sciences, 65(2), 509–523. https://doi.org/10.1007/s10620-019-05798-x
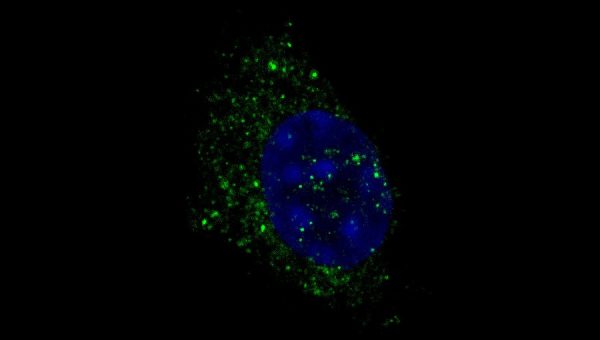
Image : Immunofluorescence des structures autophagosomes dans les myoblastes primaires dérivés de cellules souches musculaires. L’immunocoloration a été réalisée avec l’anticorps WIPI2 (vert) et les cellules ont été contre-colorées avec du DAPI (bleu).